摘要
2025年12月22日,Orphanet Journal of Rare Diseases 发表了题为《Trends, lag and characteristics of rare disease drug approval in the USA and China, 1983–2022》的研究论文,罕研社随后对其进行了中文编译。
该研究通过对1983年至2022年中美两国罕见病药物批准情况的横断面分析,系统比较了两国在孤儿药获批数量、审评时间、相关法律法规及加速计划等方面的差异。结果表明,中国持续的药品监管改革成效显著,为孤儿药审评审批工作的突破性进展提供了实证依据,揭示出监管体系的重构与改革对孤儿药研发与上市的有力推动。
背景
在过去约 40 年间,中国药品监管体系历经多项重大改革,旨在加快药物审批进程,与全球、尤其是发达国家的药物科技创新步伐保持同步。2018 年和 2023 年,国家卫健委联合多部门先后发布《第一批罕见病目录》与《第二批罕见病目录》。但目前,针对中国孤儿药获批的整体概况及审批效率的相关研究尚较为欠缺。
方法
本研究通过官方药品检索数据库,对 1983—2022 年中美两国获批的罕见病药物开展横断面分析,系统对比并剖析两国罕见病药物的获批数量、审批时长、相关法律法规及优先审评审批程序等内容。
结果
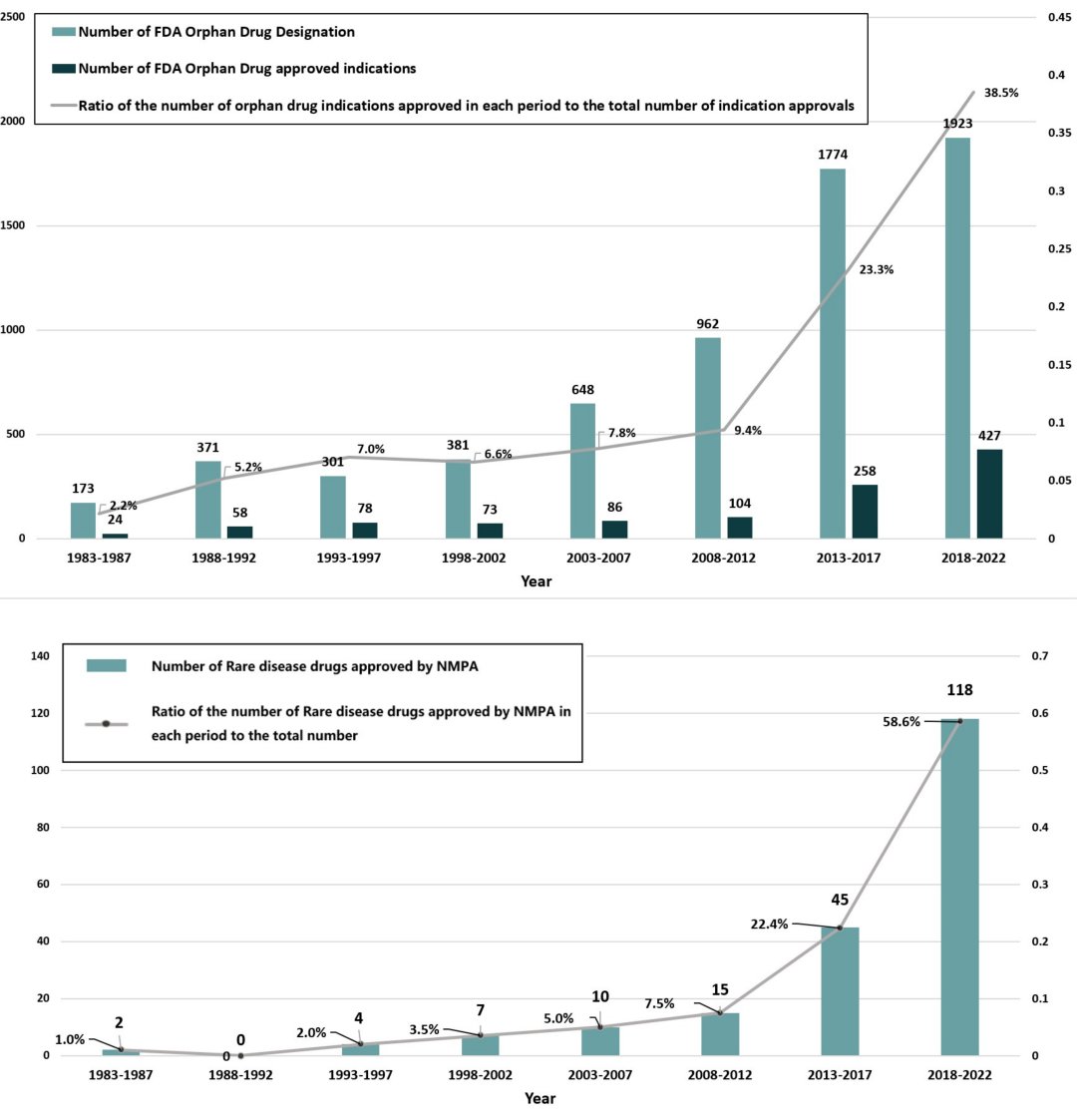
1983—2022 年间,美国食品药品监督管理局(FDA)共批准 693 种孤儿药(涵盖 1228 个剂型 / 规格)。其中,经中国国家药品监督管理局(NMPA)批准的药物共 201 种(425 个剂型 / 规格),分别占美国获批药物总数的 29.0%(201/693)、获批剂型 / 规格总数的 34.6%(425/1228)。
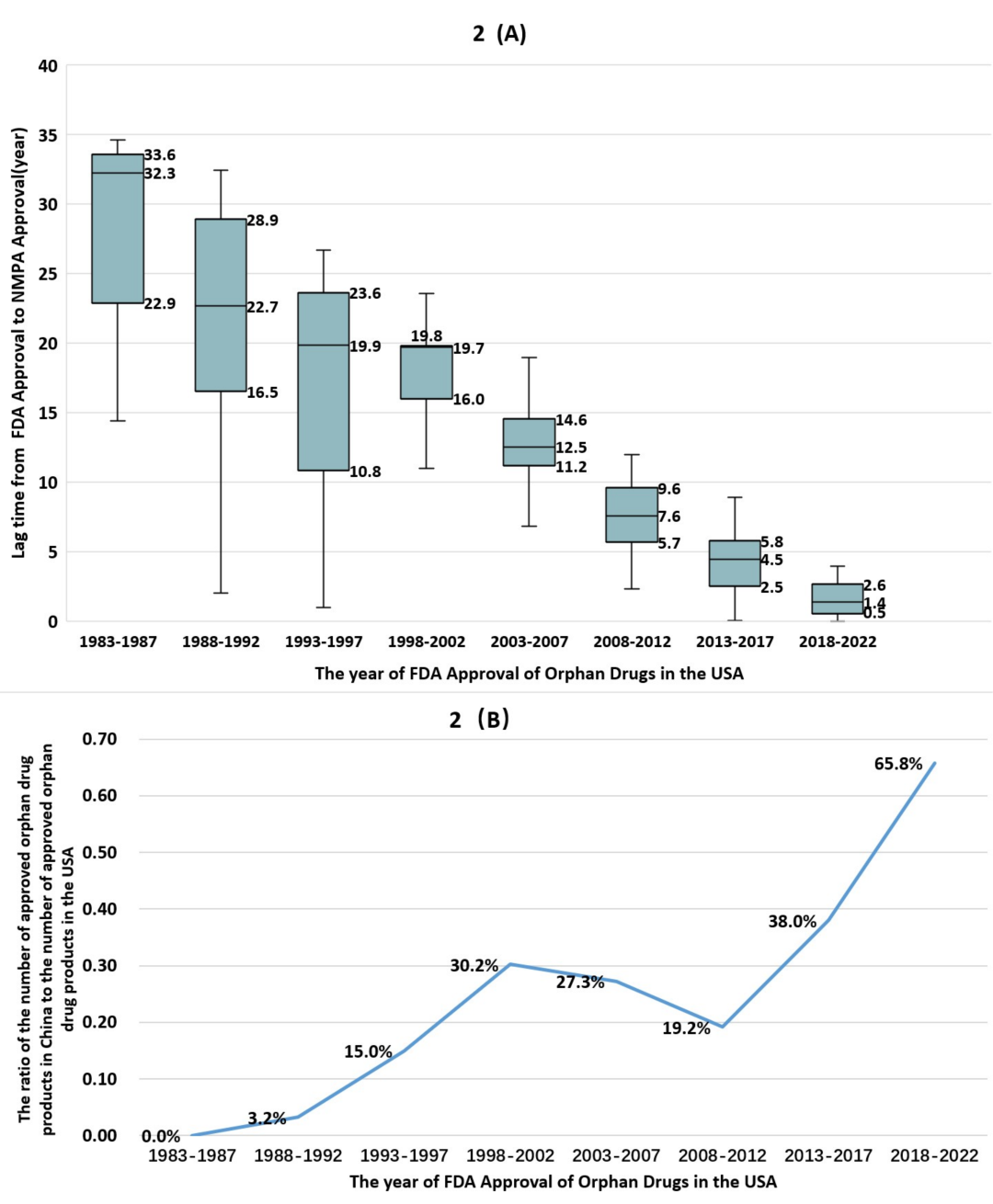
中国孤儿药获批数量呈逐年递增趋势,审批速度亦逐步加快。1983—1987 年,中美两国孤儿药审批的中位滞后时间长达 32.3 年(四分位距:22.9–33.6);而在 2018—2022 年期间,该中位滞后时间缩短至约 1.4 年(四分位距:0.5–2.6),降幅达 94.2%,这一数据印证了中国持续推进的药品监管改革取得显著成效。药物审批特殊程序对孤儿药的获批具有显著的促进作用。
结论
本研究为中国孤儿药审评审批工作取得的突破性进展提供了实证依据,揭示了中国药品监管体系的重构与改革对孤儿药研发及审批的巨大推动作用,同时也为中国未来孤儿药的发展提供了新的动力。
Part.01研究背景
孤儿药,又称罕见病药物,是用于罕见病治疗、预防及诊断的一类药物1 。不同国家对罕见病的界定标准存在差异2,3:
美国将年患病人数少于 20 万人的疾病定义为罕见病;
日本规定患病人数低于 5 万人或患病率低于 1/2500 的疾病属于罕见病;
欧盟的罕见病界定阈值为患病率不超过 5/10000。
与之不同,中国目前采用目录式管理模式界定罕见病。《第一批罕见病目录》于 2018 年 5 月 11 日发布,纳入 121 种疾病8 ;2023 年 9 月 18 日发布的《第二批罕见病目录》新增 86 种罕见病9。该目录有望在未来定期更新,纳入更多罕见病病种。
过去五年间,中国孤儿药研发进程显著加快,在研药物数量年均增长率达 34%,较全球平均增长率高出约 42%10。尽管中国已出台官方罕见病目录,但尚未发布国家级的孤儿药官方认定清单。鉴于此,本研究通过分析美国获批孤儿药在中国的可及性,并对比两国药物审批时长的差异展开研究。
美国建立罕见病相关法律框架的时间早于中国,且两国在孤儿药认定的监管体系上存在显著差异。美国 FDA 设立的特殊认定通道,能够缩短用于治疗存在未被满足医疗需求的罕见重症新药的临床研发周期及审批时长11。基于上述背景,本研究重点探讨两个核心问题:
(1)美国获批的孤儿药是否也已在中国获批上市?
(2)若已获批,其在中国的审批滞后时长为多久?
通过对这两个问题的研究,本研究旨在阐明 1983—2022 年这 40 年间中国罕见病药物监管体系的发展进程。
从历史来看,药物审批滞后曾是中国面临的突出问题12,13,这主要源于药品注册申请积压严重、监管审评周期冗长以及临床试验审批体系效率低下。
为解决这一问题,中国自 2015 年起启动了一系列药品监管改革,核心目标是通过清理积压的逾期注册申请,建立高效的审评审批流程14。后续出台的政策措施包括:扩充审评人员队伍、对新药临床试验申请(IND)设定 60 日默示许可审批时限、增设优先审评审批通道、允许进口药物同步开展国际多中心 Ⅰ 期临床试验、将国际多中心临床试验(MRCT)数据纳入中国药品审批的依据范畴15。
这些改革举措,体现出中国正逐步构建一套更为规范、全面的罕见病管理体系 15-18。改革成效显著,自 2019 年起,新药注册申请得以在法定时限内完成审批,其中优先审评审批通道将药品审批滞后时长缩短约 30 个月。尽管 2016 年以来,临床试验申请数量大幅增长,但针对审批滞后问题的细化分析仍较为匮乏14。此外,改革带来的即时积极成效,可能掩盖了导致药物审批滞后的深层次、长期性原因。
本研究旨在调查 1983—2022 年间经美国 FDA 认定并获批的孤儿药在中国的审批现状,分析审批滞后的程度,对比中美两国的特殊药物监管通道,并全面梳理当前中国孤儿药审批的整体格局。通过梳理上述发展脉络,本研究结果将凸显中国药品监管改革政策取得的切实成效,同时为中国制药企业、监管机构及药物研发部门提供实证参考依据。
讨论Part.02
中国国家药品监督管理局孤儿药获批数量显著增长
在过去四十年间(1983-2022 年),经美国 FDA 获批后又在中国获批的孤儿药数量持续增加,且中美两国的审批时间差逐步缩短。这一进展与中国多项优先审评审批通道的建立及完善进程相契合。
截至目前,仅有三项已发表研究专门聚焦于中国与其他国家之间的药物审批滞后问题 23-25。本研究将药品监管机构对罕见病药物的审评审批效率,作为衡量监管能力与水平的核心指标。明确审批滞后的特征并找出其关键驱动因素,可为中国政府制定更具针对性的政策措施、缩小药物可及性差距提供参考依据。
在 “健康中国” 战略的指导下,国家对罕见病防治工作的重视程度日益提升,先后出台一系列政策文件,推动罕见病药物研发,改善罕见病患者的治疗可及性与医疗保障水平。核心举措包括:
(1) 清理积压的逾期注册申请,建立高效的审评审批流程27。
(2) 设立优先审评审批通道,可将药物审批滞后时长缩短约 30 个月 20,23。
(3) 放宽进口药物早期研发相关限制,鼓励参与全球药物研发工作 28,29。
(4) 赋予所有进口罕见病药物优先审评资格,法定审评时限仅为 10 个工作日14。
此外,这一轮改革推动中国在 2019 年对《药品管理法》进行了全面修订,并重新制定了多项重要配套法规 30。国际人用药品注册技术协调会(ICH)大会接纳国家药品监督管理局(NMPA)成为监管机构成员,这一里程碑事件标志着中国在药品监管领域已跻身全球行列。
其他重要举措包括:2019 年,国家卫生健康委员会牵头组建了国家罕见病诊疗协作网;2018 至 2020 年,国家药监局发布了三批《临床急需境外新药名单》,共纳入 73 种药物,其中罕见病治疗药物 33 种。该名单重点收录了已在美国、欧盟或日本获批,但尚未在国内上市,且用于治疗存在未被满足医疗需求的严重疾病的新药,名单内药物可享受优先审评审批资格。2021 年,《罕见疾病药物临床研发技术指导原则》正式发布,进一步提升了罕见病药物的临床研发效率。
上述一系列协同推进的举措,在一定程度上解释了近年来中国罕见病药物审批时长为何能逐步向美国看齐。本团队开展的一项相关研究显示,中国罕见病药物的可及性呈稳步上升趋势:截至 2020 年,医院端药物可及率中位数达到 41.1%,高可及性药物数量增幅达 16.5%;此外,在 74 种孤儿药中,已有 64 种被纳入国家基本医疗保险体系,城乡居民可享受医保报销,纳入比例较之前提升了 14.1%31。
尽管中美两国在政治体制、经济发展水平及文化背景上存在差异,但美国 FDA 的孤儿药认定机制仍为中国提供了宝贵的借鉴经验。清晰且标准化的孤儿药定义,是开展孤儿药认定工作的基础前提32-34。
一项在中国开展的访谈调研显示,78.6% 的受访者(罕见病患者组织代表)支持为罕见病药物研发提供激励政策,71.4% 的受访者建议加快出台孤儿药相关专项立法
35。因此,基于对美国及其他国家孤儿药认定机制的研究36,本研究建议:中国应结合本国国情,基于流行病学数据、临床疗效及卫生经济学指标,建立罕见病与孤儿药的正式定义。这一定义的明确,将为后续制定孤儿药研发激励政策奠定坚实基础38,39,同时也能为完善中国罕见病患者的治疗与保障体系提供法律依据。
本研究存在一定局限性:国家药监局未公开药物研发企业提交注册申请的具体时间,仅公示了最终获批日期。审评时限相关信息的透明度不足,导致研究无法对审批流程开展更精细化的分析,这也是未来研究需重点关注的方向。
推动美国 FDA 孤儿药认定与获批数量显著增长的因素
过去 40 年间,美国 FDA 获批的孤儿药总数大幅攀升。在过去 5 年里,孤儿药在 FDA 批准的新分子实体中占据了相当大的比例40。这一成果是多种因素共同作用的结果4,40-44:
(1)政策与指南因素:1983 年 1 月颁布的《孤儿药法案》,后续分别于 1984 年、1985 年、1988 年及 2017 年历经修订,构建起了孤儿药研发的激励框架基础。2002 年出台的《罕见病法案》进一步提供了额外的研究资金与法律支持,对孤儿药研发形成了有力支撑。2017 年 6 月 FDA 推出的孤儿药现代化计划成为关键推动因素,该计划承诺在 90 天内处理完所有积压的孤儿药认定申请,并确保所有新提交的申请在 90 天内得到反馈。这项政策极大地提升了制药行业参与孤儿药研发的积极性。目前实施的激励政策包括:临床试验费用享受 25% 的税收抵免(此前比例为 50%)、豁免新药上市申请(NDA)和生物制品许可申请(BLA)的相关费用、获得研究资助的资格、适用优先审评审批通道、豁免部分临床数据要求,以及获批后享有 7 年的市场独占权。
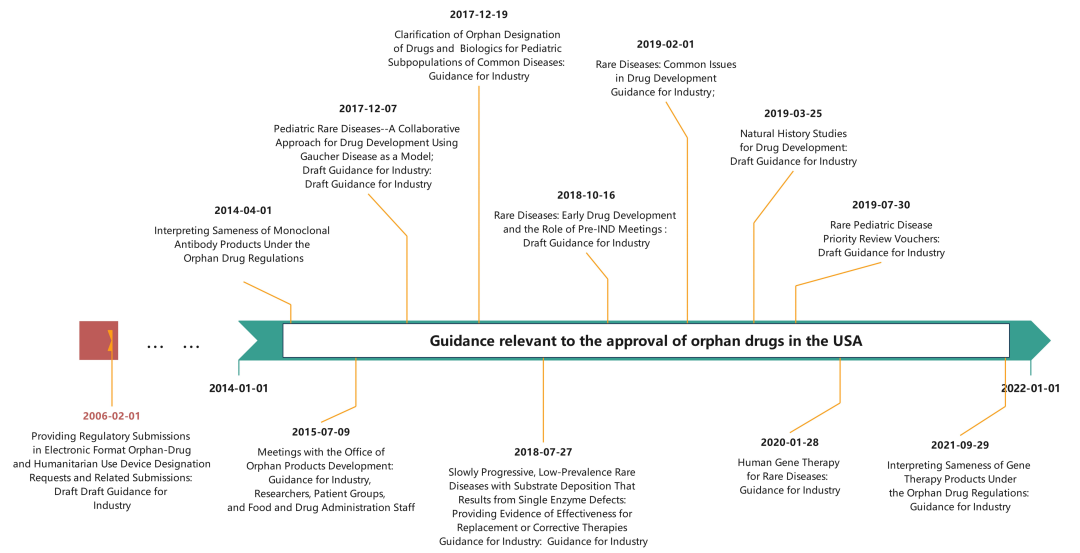
(2)技术因素:约 80% 的罕见病属于遗传性疾病。基因组学、基因测序、分子生物学及生物技术领域的突破性进展,实现了药物靶点的精准定位,显著提升了制药企业的研发能力。这使得生物制品、罕见癌治疗药物及靶向治疗药物的获批数量出现明显增长。
(3)商业激励因素:溢价定价与受保障的市场独占权等商业激励措施,能为制药企业带来高额回报,有力地刺激了孤儿药的研发进程28。有研究表明,拥有孤儿药获批资质的企业,其投资回报率较同行业其他企业高出 9.6%45。《2019 年爱唯医药孤儿药报告》(Evaluate Pharma Orphan Drug Report 2019)证实,孤儿药在美国市场的定价远高于普通药物。但这类高价策略,以及由市场独占权所形成的垄断格局 —— 可能会抑制市场竞争 —— 至今仍是业内激烈争论和热议的话题46-50。



















