摘要
原发性线粒体病(primary mitochondrial diseases, PMD)是一类由线粒体功能障碍引发、病种数量持续增长的疾病。目前,关于 PMD 的精准定义尚未形成共识。
为此,本研究提出一套分类体系构建方案,旨在实现对 PMD 的标准化、系统化分类,且该体系与国际线粒体病分类数据库(ICIMD) 及先天性代谢缺陷数据库(IEMBase) 保持一致。
本研究共纳入 452 个 PMD 致病基因,将疾病划分为 18 个类别,具体如下:
氨基酸代谢障碍 (Disorders of amino acid metabolism)
肽类及胺类代谢障碍 (Disorders of peptide and amine metabolism)
碳水化合物代谢障碍(Disorders of carbohydrate metabolism)
脂肪酸及酮体代谢障碍 (Disorders of fatty acid and ketone body metabolism)
能量底物代谢障碍 (Disorders of energy substrate metabolism)
线粒体 DNA 相关疾病 (Mitochondrial DNA-related disorders)
核基因编码的氧化磷酸化障碍(Nuclear-encoded disorders of oxidative phosphorylation)
线粒体辅因子生物合成障碍 (Disorders of mitochondrial cofactor biosynthesis)
线粒体 DNA 维持与复制障碍 (Disorders of mitochondrial DNA maintenance and replication)
线粒体基因表达障碍(Disorders mitochondrial gene expression)
其他线粒体功能障碍(Other disorders of mitochondrial function)
代谢物修复 / 校正障碍 (Disorders of metabolite repair/proofreading)
脂质代谢障碍(Disorders of lipid metabolism)
碱基、核苷酸及核酸代谢障碍(Disorders of nucleobase, nucleotide and nucleic acid metabolism)
四吡咯代谢障碍(Disorders of tetrapyrrole metabolism)
细胞器生物发生、动态变化及互作障碍(Disorder of organelle biogenesis, dynamics and interaction)
维生素及辅因子代谢障碍(Disorders of vitamin and cofactor metabolism)
神经递质相关障碍(Neurotransmitter disorders)
同时,本研究还阐述了该病累及的 22 个器官系统的临床特征及实验室检查特点。按致病基因统计,最常见的受累症状依次为神经系统症状(21.1%)、眼部症状(10.3%)、肌肉症状(9.0%)、胃肠道症状(8.3%)及心血管系统症状(7.9%)。
1 引言
原发性线粒体病(PMD)是一类由线粒体功能障碍引发、病种数量持续增长的疾病 [1]。这类疾病具有多维度异质性特征,具体表现为:(1)临床异质性,涵盖发病年龄差异、单器官或多器官受累的区别,以及受累组织类型的不同;(2)生化异质性,迄今尚未发现兼具高灵敏度与高特异性的生物标志物(这一现象很可能与线粒体的多重生理功能相关 —— 线粒体的任一功能受损均可能引发 PMD);(3)遗传异质性,线粒体的功能受核基因组与线粒体基因组双重调控,大量核基因的变异均与 PMD 相关,同时该病还存在线粒体 DNA(mtDNA)母系遗传、新发突变及异质性等遗传特点。
基因组学时代的到来,推动 PMD 相关致病基因的数量快速增长,同时也加深了人们对疾病潜在发病机制的理解。此外,随着基因检测技术的普及,意义未明变异(VUS)的检出数量日益增多,这类变异需通过功能验证才能被判定为(疑似)致病性变异。
线粒体医学仍是一门新兴学科,目前仍存在诸多基础性问题亟待解决,其中就包括 PMD 的精准定义尚未形成共识。已有研究将 PMD 定义为:由基因突变直接或间接引发氧化磷酸化(OXPHOS)系统功能障碍,或导致线粒体结构与功能出现其他异常的疾病,这些异常包括线粒体超微结构紊乱、辅因子与维生素合成障碍,以及线粒体内部其他代谢通路(如三羧酸循环、丙酮酸代谢)受损 [1]。
本研究旨在填补当前认知空白,提出一套用于 PMD 系统化分类的疾病分类体系。研究将上述疾病定义列为首要纳入标准,同时纳入另外两项标准,分别与编码蛋白或酶的亚细胞定位相关,以及与非线粒体定位蛋白或酶缺失对线粒体功能造成的影响相关。
在新一代测序技术广泛应用的当下,一套清晰合理的疾病分类体系至关重要,可指导临床在诊断检测中合理纳入相关基因 —— 尤其是在基因检测手段已逐步从传统靶向基因组合检测,向全外显子测序与全基因组测序转变的趋势下。将一套全面且标准化的 PMD 分类体系,应用于基因检测报告解读及专科转诊流程中,有助于确保患者获得及时准确的诊断,从而缩短该领域长期存在的诊断延误周期。
对致病基因进行系统性分类,同样有助于深入解析疾病发病机制,或可为这类现有疗法基本难以根治的难治性疾病,开辟全新的治疗思路。此外,本研究通过与先天性代谢缺陷数据库(IEMbase)[2] 进行关联分析,阐述了 PMD 的核心临床特征。
在相关知识库快速扩充的背景下,PMD 的明确定义及清晰的纳入排除标准,其重要性不言而喻。这不仅对患者及其家属意义重大,同时也影响着照护这类患者的医护人员,以及为患者提供医疗服务与资源的整个医疗体系 [3-7]。
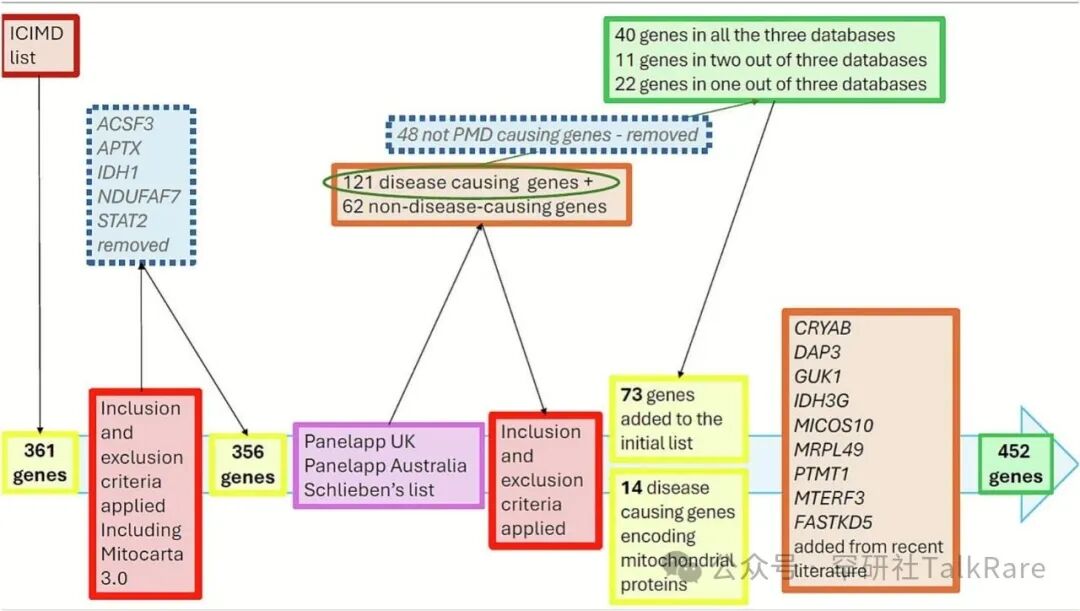
图 1 线粒体特征基因集构建流程。研究首先以国际线粒体病分类数据库(ICIMD)中的初始原发性线粒体病(PMD)基因集为基础,依据正文中所述的纳入与排除标准进行筛选,剔除 5 个不符合条件的基因(详见正文)。随后,将筛选后剩余的基因与另外 3 个临床应用基因集进行比对,再次执行纳入与排除标准,最终新增 73 个 PMD 致病基因。在此基础上,补充纳入 14 个编码线粒体蛋白、但未被常规归类为 PMD 的致病基因。最后,经文献调研进一步增补 9 个基因,最终构建得到包含452 个 PMD 致病基因的基因集。
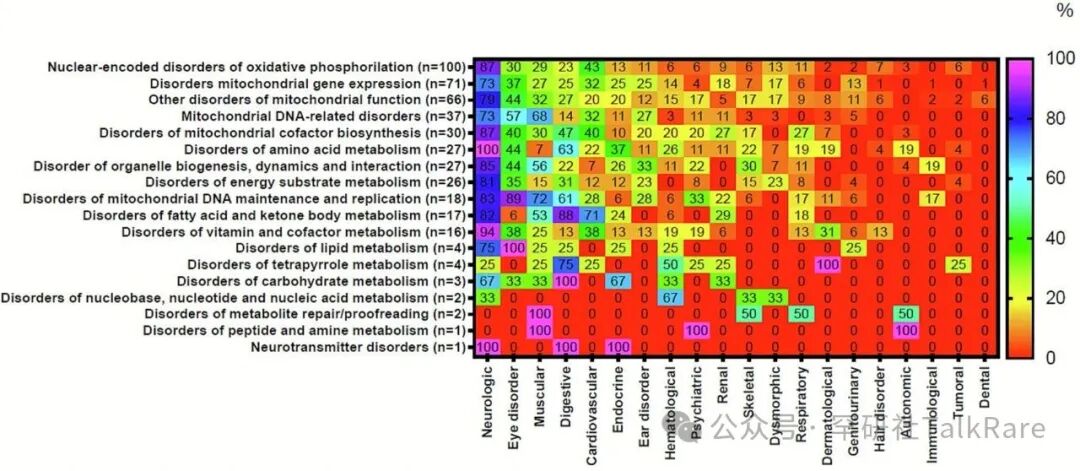
图 2 十八类原发性线粒体病(PMD)中 20 个受累器官 / 系统的发生频率(%)分布热图。注:器官 / 系统按受累频率从高到低依次列于横轴,18 类 PMD 按发病率从高到低依次列于纵轴。统计分析的计数基准为单个基因缺陷,而非受累患者总数。各器官 / 系统受累频率的计算方式为:以某一疾病类别中存在任意器官 / 系统受累表现的疾病总数作为分母,统计该类别中累及特定器官 / 系统的疾病数量占比。热图颜色梯度定义如下:红色代表该疾病类别中无相关症状报道(发生率 0%),紫色代表该症状在对应疾病类别中发生频率最高(发生率 100%)。详细数据信息参见补充表 S1 及 S2。(关于本图例中颜色标注的具体说明,请参阅本文电子版。)
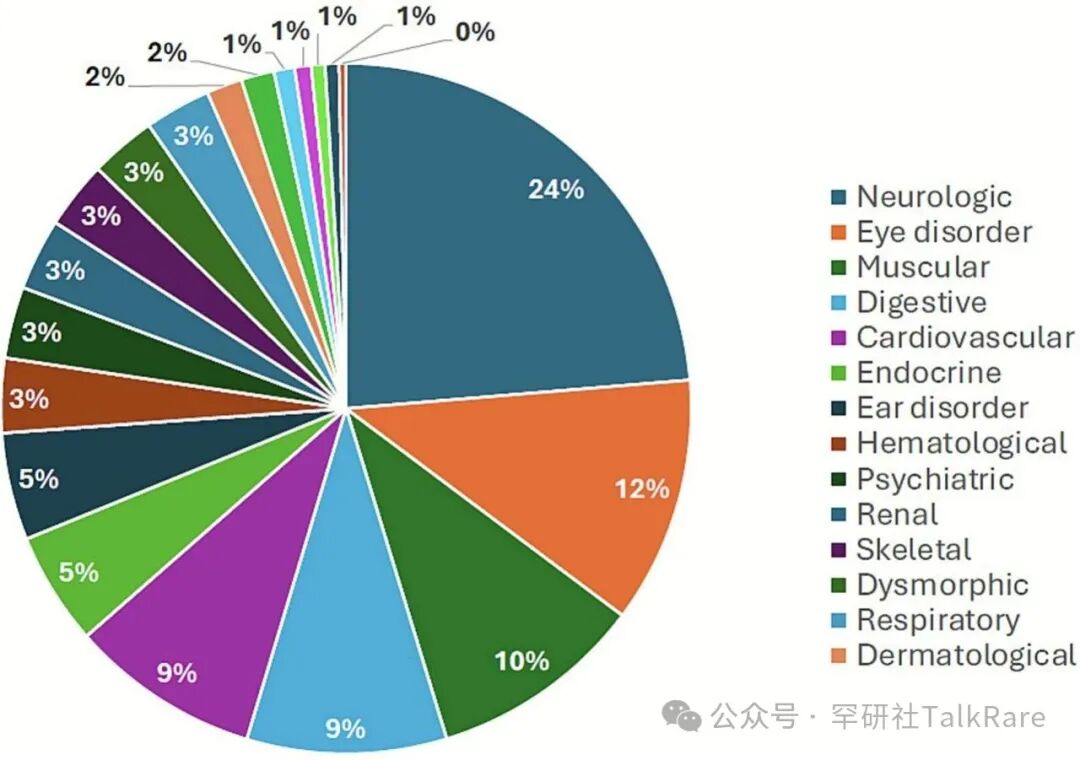
图 3 452 种原发性线粒体病(PMD)的症状发生频率(%)分布注:症状按受累器官 / 系统归为 20 大类,详细数据参见表 1。
4 讨论
4.1 命名与分类体系的重要性
制定一套清晰且尽可能获得公认的 PMD 定义,其意义并非可有可无,更不仅局限于学术层面。该定义对患者及其家属具有切实价值 —— 凭借明确的疾病界定,患者能够获得针对性的专科诊疗服务 [3,6]。此外,严谨的疾病分类体系可助力确定临床试验的精准研究人群,使患者获得参与试验性治疗的机会 [22]。明确疾病的定义、特征及发病机制,有助于设定清晰的临床试验终点,进而提升治疗方案获得监管机构批准的可能性。
另一重要意义在于,明确的疾病实体界定能够帮助患者更顺利地获取经济与社会支持,对接疾病相关的患者权益倡导组织,而这些资源对于存在复杂需求的患者及家属而言,均是至关重要的 [23]。
疾病定义不仅对患者群体意义重大,也对临床医生的诊疗工作产生深远影响:借助规范化的疾病定义,临床医生能够为患者提供更优质、更具靶向性的治疗方案,并向相关领域专家寻求诊疗支持 [24]。最后,在许多国家,医疗费用的报销政策均以疾病编码与分类体系为依据制定 [25]。
(本文仅转译了部分内容,如需阅读全文请阅读原文)
本文作者来自伦敦大学学院大奥蒙德街儿童健康研究所。图片、文章版权均属于原作者所有,如有侵权,请及时告知,我们会在24小时内删除相关信息。欢迎转载,转载请注明出处。



















