文章来源:中华神经科杂志, 2025, 58(06): 624-631.
作者:许小菁 王婷 程苗苗 欧阳世佳 杨莹 杨小玲 刘昌昊 张月华
摘要
目的
总结编码钠离子通道β1亚基的SCN1B基因相关癫痫患儿基因型和临床表型特点。
方法
在2016年5月至2024年7月于北京大学第一医院儿童医学中心就诊的疑似遗传性癫痫的患儿中,以二代测序技术发现SCN1B变异并经过Sanger测序或荧光定量聚合酶链反应方法验证的癫痫患儿为研究对象进行基因型和临床表型分析。
结果
共纳入17例患儿,男性8例,女性9例;携带SCN1B错义变异10例(2例携带相同变异),缺失变异4例,无义变异、剪切位点变异及外显子4~5缺失各1例;6例为SCN1B新生变异,11例为遗传自父母一方的变异。11种变异位点尚未见报道。癫痫发作起病年龄为3月龄至5岁3月龄(中位年龄:14月龄)。癫痫发作类型包括全面强直阵挛发作(GTCS)14例,局灶性发作9例,肌阵挛发作3例,不典型失神2例,癫痫性痉挛、强直发作及失张力发作各1例。在17例患儿中,11例有多种发作类型,14例病程中有热敏感特点。脑电图显示局灶性放电3例,局灶性及广泛性放电3例,多灶性放电6例;4例监测到临床发作,其中肌阵挛发作1例、GTCS 1例、不典型失神发作1例、监测到肌阵挛发作和强直发作1例。诊断符合遗传性癫痫伴热性惊厥附加症9例(9/17),婴儿肌阵挛癫痫和婴儿癫痫性痉挛综合征各1例,非特异性发育性癫痫性脑病2例,其余4例不能诊断为癫痫综合征。治疗有效的抗癫痫发作药物(ASM)包括丙戊酸(8例)、左乙拉西坦(5例)、托吡酯(3例)、氯巴占(2例)、氯硝西泮及氨己烯酸(1例)。钠通道阻滞剂引起发作加重3例(奥卡西平引起发作加重2例、拉莫三嗪引起发作加重1例)。末次随访时14例(14/17)发作控制半年及以上,3例尝试2种以上ASM发作仍未控制。13例发育正常,4例发育落后。
结论
SCN1B基因相关癫痫患儿的基因杂合变异包括错义、缺失、无义、剪切位点变异及外显子缺失,不同基因变异与临床表型之间的相关性不明确。SCN1B基因相关癫痫患儿发病年龄从婴儿期至学龄前期,可表现多种发作类型,以GTCS最常见,表型轻重变化大,可表现为预后良好的热性惊厥或热性惊厥附加症,也可表现为发育性癫痫性脑病。ASM中以丙戊酸有效率最高,钠通道阻滞剂可加重部分患儿的发作,应用需谨慎。
电压门控钠通道是由4个α亚基和2个β调节亚基组成的大型跨膜蛋白[1]。单独的α亚基可以显示功能性通道特性,但需要β亚基来调节钠通道失活[2]。不同的β亚基由SCN1B、SCN2B、SCN3B、SCN4B 4种基因编码。其中SCN1B基因编码β1亚基,在大脑和心脏均有表达[3]。β1亚基含有一个细胞外Ig结构域、跨膜结构域以及具有多个磷酸化位点和分泌酶裂解位点的细胞内结构域。目前已有报道少数SCN1B基因纯合变异可导致类似Dravet综合征的发育性癫痫性脑病(developmental epileptic encephalopathy,DEE),而杂合变异与热性惊厥(febrile seizures)、遗传性癫痫伴热性惊厥附加症(genetic epilepsy with febrile seizures plus,GEFS+)、颞叶癫痫、特发性全面性癫痫有关[4, 5, 6, 7]。SCN1B基因变异具有外显率不全的特点,也可能参与良性家族性婴儿癫痫的发病[8]。此外,SCN1B基因变异还可能与心律失常的相关疾病如Brugada综合征有关[9]。
我们收集北京大学第一医院儿童医学中心儿童神经内科就诊的SCN1B基因杂合变异相关癫痫患儿资料,总结其临床表型及基因变异特点,并对患儿预后进行随访,以提高临床医师对该基因变异相关癫痫的认识。
资料和方法
一、研究对象
回顾性收集2016年5月至2024年7月在北京大学第一医院儿童医学中心儿童神经内科门诊就诊的已通过二代测序技术明确癫痫致病基因、诊断为遗传性癫痫的患儿,共3 445例。筛选出携带SCN1B基因杂合变异的17例(17/3 445,0.49%)癫痫患儿为研究对象,总结其临床表型和基因变异特点,并对患儿的治疗和预后进行随访。对所有患儿均建立了临床资料登记表,内容包括姓名、性别、出生日期、民族、发病年龄、癫痫发作表现、围生期情况、智力运动发育情况、家族史、用药史,以及脑电图、头颅磁共振成像(magnetic resonance imaging,MRI)等辅助检查结果。对患儿进行定期门诊复诊或电话随访。
本研究为回顾性病例系列研究,获得了北京大学第一医院伦理委员会批准(批准文号:2012[453]),并获得了患儿家长的书面知情同意。
二、基因检测方法
采集先证者和家系成员的外周血,提取DNA进行家系全外显子组测序,SCN1B基因转录本参考国际标准转录本ENST00000262631(NM_001037)。对于测序发现的可疑SCN1B基因致病性单核苷酸变异者,进一步采用Sanger测序验证变异位点,明确变异来源、进行家系共分离分析。对于存在SCN1B基因致病性拷贝数变异者,以ALB基因为内参基因,采用荧光定量聚合酶链反应方法进行扩增,进行先证者及家系成员的验证。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)与分子病理学会(Association for Molecular Pathology,AMP)联合共识推荐指南[10]对变异的致病性进行评估,通过Mutation Taster(http://www.mutationtaster.org/)、PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/PolyPhen2)和SIFT(http://provean.jcvi.org/index.php)网站预测致病性。
结果
一、患儿基本资料
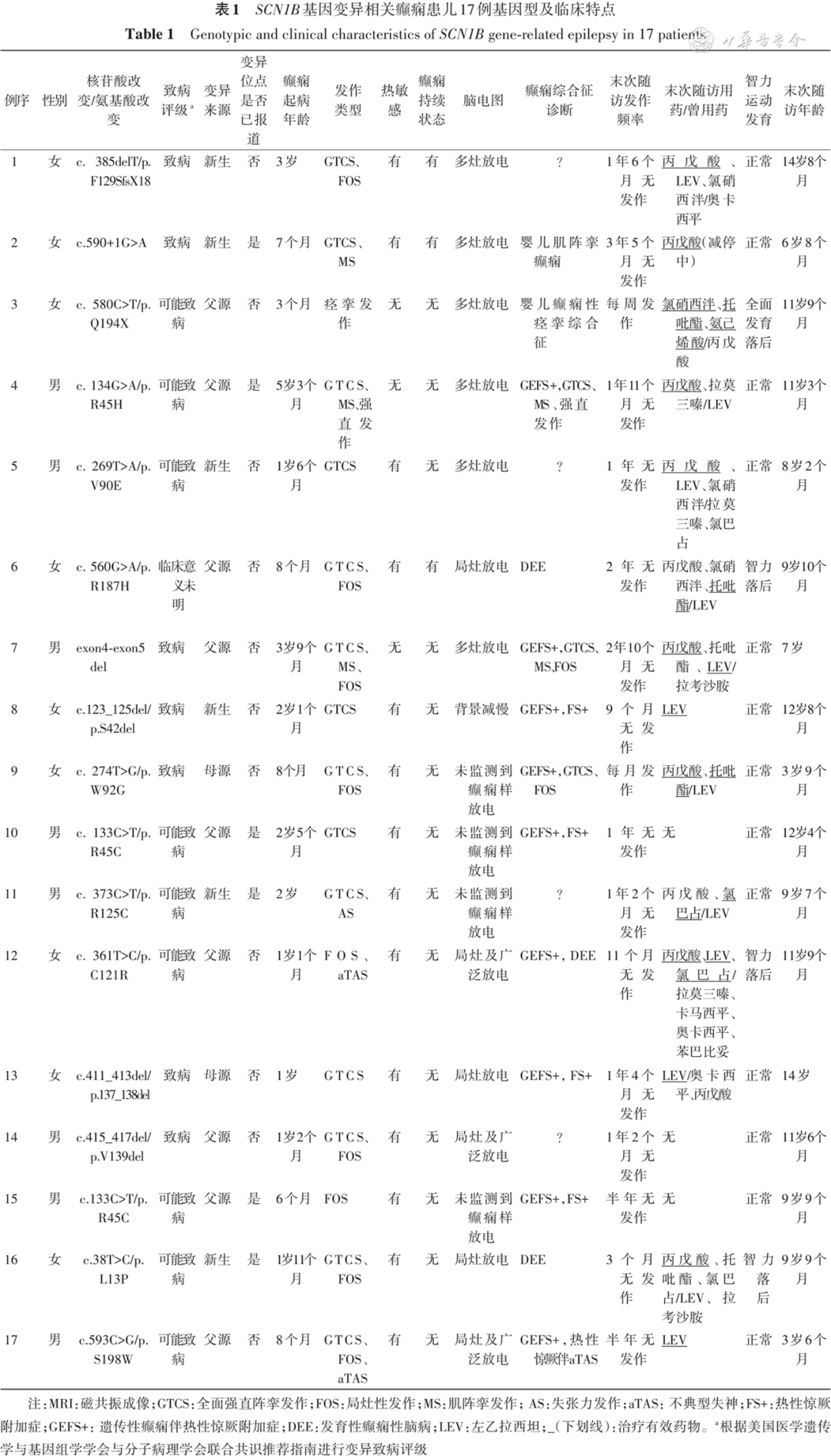
共收集了17例患儿,其中男性8例、女性9例。9例患儿(例4、例7、例8、例9、例10、例12、例13、例15、例17)有癫痫或热性惊厥家族史,可归为GEFS+,其家系成员表现正常或热性惊厥、热性惊厥附加症(febrile seizures plus,FS+)等。具体9个家系成员表型及SCN1B变异结果见图1。17例患儿SCN1B基因变异及临床特点见表1。
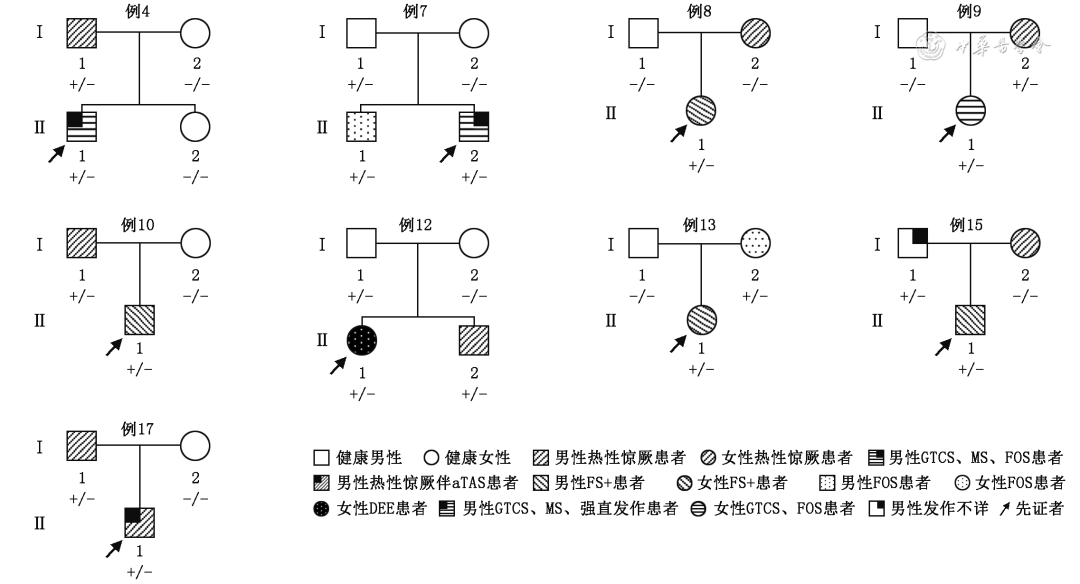
FS+:热性惊厥附加症;GTCS:全面强直阵挛发作;FOS:局灶性发作;MS:肌阵挛发作;aTAS:不典型失神发作;DEE:发育性癫痫性脑病
图1 9个遗传性癫痫伴热性惊厥附加症家系临床表型及SCN1B基因变异
Figure 1 Clinical phenotypes and SCN1B gene variants in 9 families with genetic epilepsy with febrile seizures plus
二、基因检测结果
通过全外显子组测序,17例患儿均携带SCN1B基因杂合变异,其中携带9种错义变异10例(R45H、V90E、R187H、W92G、R45C、R125C、C121R、L13P、S198W)、缺失变异4例(c.385delT/p.F129SfsX18、c.123_125del/p.S42del、c.411_413del/p.137_138del)
c.415_417del/p.V139del无义变异1例(c.580C>T/p.Q194X)、剪切位点变异1例(c.590+1G>A)、外显子4~5缺失1例(exon 4-exon 5 del)。在17例患儿中,6例为SCN1B基因新生变异,9例为遗传自父亲的基因变异,2例为遗传自母亲的基因变异。11种基因变异未见文献报道及ClinVar数据库(https://www.ncbi.nlm.nih.gov/clinvar/)收录。例6患儿携带变异根据ACMG和AMP联合共识推荐指南[10]判定为临床意义未明,通过Mutation Taster、PolyPhen-2和SIFT软件预测结果为有害。
三、临床表现
17例患儿的癫痫发作起病年龄为出生后3个月至5岁3个月,中位起病年龄为出生后1岁2个月。癫痫发作类型包括全面强直阵挛发作(generalized tonic-clonic seizure,GTCS)14例,局灶性发作9例,肌阵挛发作3例,不典型失神发作2例,癫痫性痉挛、强直发作及失张力发作各1例。11例患儿病程中有2种以上发作类型,6例患儿病程中仅有1种发作类型包括癫痫性痉挛(例3)、GTCS(例5、例8、例10、例13)、局灶性发作(例15)。3例患儿病程中曾出现发作大于30 min的癫痫持续状态。14例患儿发作有热敏感特点,发热易诱发发作。癫痫综合征诊断包括可归为GEFS+9例、婴儿肌阵挛癫痫1例、婴儿癫痫性痉挛综合征1例、非特异性DEE 2例,余4例患儿不能诊断为癫痫综合征。归为GEFS+的患儿家系中先证者表型包括1例非特异性DEE(例12),家系成员表型包括热性惊厥(例4、例10及例17患儿父亲,例8、例9及例15患儿母亲,例12患儿同胞弟弟)、伴或不伴发热的局灶性发作(例7患儿同胞哥哥、例13患儿母亲),发作不详(例15患儿父亲)。具体见图1。
四、辅助检查
1.脑电图:对本组患儿均行4 h以上视频脑电图监测,发现背景减慢1例;发作间期有局灶性放电3例、局灶性及广泛性放电3例,多灶性放电6例,4例发作间期未监测到癫痫样放电。4例监测到临床发作,其中肌阵挛发作1例(例2)、GTCS 1例(例7)、不典型失神发作1例(例12)、监测到肌阵挛发作和强直发作1例(例4)。
2.头颅MRI:例2患儿头颅MRI提示左颞脉络膜囊肿;例11患儿病程中曾有GTCS,持续7 min,头颅MRI提示轻度脑萎缩;余14例患儿均正常。
五、治疗和预后随访
16例患儿末次随访年龄为3岁6个月至14岁8个月,治疗有效的抗癫痫发作药物(anti-seizure medications,ASM)包括丙戊酸(8例)、左乙拉西坦(5例)、托吡酯(3例)、氯巴占(2例)、氯硝西泮及氨己烯酸(1例)。14例患儿半年及以上无发作,其中目前使用丙戊酸8例、左乙拉西坦7例、氯硝西泮3例、托吡酯及氯巴占各2例、拉莫三嗪1例,3例(例10,例14和例15)未使用ASM。本组长期规律使用ASM的患儿中,4例服用1种药物(例2服用丙戊酸,例8、例13及例17服用左乙拉西坦),10例服用2种或2种以上药物。从药物保留率上看丙戊酸最高。本组中10例患儿均长期使用丙戊酸,其中8例患儿应用有效。钠通道阻滞剂引起发作加重3例,其中奥卡西平引起发作加重2例(例1和例13)、拉莫三嗪引起发作加重1例(例5)。在既往用药中,例3患儿使用丙戊酸无效,例7及例16患儿使用拉考沙胺无效,4例患儿(例6、例9、例11、例16)使用左乙拉西坦无效。本组患儿中末次随访时4例患儿发育落后,其中3例智力落后,1例全面发育落后,余患儿发育正常。
讨论
SCN1B基因定位于染色体19q13.11,含5个外显子,编码钠通道β1多功能辅助亚基,调节通道门控和通道表达水平[7,11]。蛋白质β1亚基作为跨膜蛋白,与细胞黏附蛋白相互作用[12]。作为癫痫的致病基因之一,SCN1B基因的杂合变异和纯合变异均可导致癫痫,杂合变异有外显率不全的特点。基因变异类型包括错义、无义、框移、剪切位点及片段基因缺失或重复。1998年Wallace等[13]首次报道了SCN1B基因杂合变异(p.C121W)与GEFS+有关,家系中的SCN1B基因变异携带者表现不同的表型,如热性惊厥、FS+、FS+伴失神等,也可表现正常。2002年Wallace等[14]又在另一个GEFS+家系中发现相同的SCN1B基因致病杂合变异。2009年Patino等[6]报道了第一例与SCN1B基因纯合突变(p.R125C)相关的Dravet综合征患者。后在5个家系发现3例DEE患儿携带2种SCN1B基因纯合致病变异[15]。此外在GEFS+家系中携带SCN1B基因致病杂合变异(IVS2-2A>C)也可表现为早发性失神癫痫,该变异在蛋白质水平上将导致基因表达的细胞外免疫球蛋白环结构域中的5个氨基酸缺失[16]。除癫痫发作外,SCN1B基因变异还可引起心房颤动(13型)和Brugada综合征(5型)[9,17]。本组癫痫患儿中携带SCN1B基因杂合变异类型以错义变异(10/17)为主,且多为遗传性变异(11/17)。其中5个家系中(例3、例6、例7、例12、例14)均有携带与先证者相同变异但表现正常的成员。GEFS+家系中携带SCN1B基因杂合变异者表型可为较轻的热性惊厥或FS+,也可为影响智力发育的非特异性DEE。由此可见SCN1B基因变异多为遗传性变异,常出现在GEFS+家系中,表型相对较轻,但具有外显率不全和表型异质性特点。
随着二代测序技术的临床应用,SCN1B基因变异相关癫痫的报道逐渐增多,但国内文献报道尚少。既往文献报道的杂合变异相关癫痫的起病年龄从生后2 d到9岁[5,14]。本组患儿起病年龄从婴儿期到学龄前期,同样跨度较大。在先前报道的SCN1B基因杂合变异患者中,发作类型包括GTCS、阵挛性发作、失神发作、局灶性发作、痉挛发作、肌阵挛-失张力发作和失张力发作。癫痫表型包括热性惊厥、FS+、早发失神癫痫、颞叶癫痫以及可出现癫痫性猝死在内的局灶性癫痫[5,14,16,18, 19]。在已报道的携带SCN1B基因纯合变异的患儿中,发作类型包括GTCS、局灶性发作、肌阵挛发作、失神发作和阵挛性发作,癫痫表型主要为Dravet综合征、DEE等较严重类型[15,20, 21, 22, 23]。本组患儿中癫痫发作类型同样多样,除已报道的GTCS、局灶性发作、肌阵挛发作和失张力发作,还包括强直发作、不典型失神发作和癫痫性痉挛。癫痫表型除已报道的热性惊厥、FS+,还包括未报道的婴儿肌阵挛性癫痫、婴儿癫痫性痉挛综合征、非特异性DEE。
在本研究中,在患儿病程中监测到其脑电图表现与其发作类型相关的异常放电(12/17)。既往已报道的SCN1B基因杂合变异相关癫痫患儿表现为不同的发作类型,脑电图监测到了局灶性、多灶性和全面性癫痫样放电,但不具有特异性[5,14,16,18,24]。本组患儿均完善了头颅MRI检查,均没有特异性改变。既往文献报道,可获得头颅影像学信息的8例SCN1B基因杂合变异相关癫痫患儿中,4例患儿头颅MRI正常,余4例患儿表现异常:1例热性惊厥患儿表现为脑室旁白质软化,3例颞叶癫痫患儿头颅MRI分别表现为非特异性弥漫性白质高信号、双侧海马硬化和右侧海马体积减小[5,14,24]。在12例已报道的SCN1B基因纯合变异相关癫痫患儿中,6例头颅MRI正常,另6例患儿头颅MRI出现非特异性脑萎缩、半卵圆中心周围多个小梗死伴周围胶质增生、弥漫性脑萎缩、轻度萎缩等改变[6,15,20, 21, 22, 23,25]。总体而言,SCN1B基因杂合变异相关癫痫患儿头颅MRI大多数正常,而携带SCN1B基因纯合变异的患者可能出现脑萎缩和其他非特异性变化。携带SCN1B基因杂合变异的癫痫患儿发作相对容易控制,既往文献报道的患儿使用的有效ASM有丙戊酸、氯巴占、卡马西平,也有患儿在加用苯妥英后发作得到控制[16,18,24]。本组患儿末次随访时多数发作控制较好,药物治疗多选择丙戊酸和左乙拉西坦,提示以上广谱ASM的有效率较高。
文献多次报道与癫痫相关的SCN1B基因杂合变异体有p.R125L[26, 27]和p.C121W[5,13, 14,28, 29],携带上述变异的患者表现为热性惊厥、FS+、颞叶癫痫等。位于第85个氨基酸的SCN1B基因杂合变异体可能与热性惊厥和FS+表型有关[5]。本研究中也发现了2例携带SCN1B基因变异位点R45C的患儿(例10和例15),均表现为GEFS+家系中的FS+表型,未长期使用ASM,而同一位点发生错义变异R45H的患儿(例4)表现为GTCS、肌阵挛和强直发作,规律服用2种ASM。诊断为癫痫性脑病(婴儿癫痫性痉挛综合征、非特异性DEE)的患儿(例3、例6、例12和例16)携带的SCN1B基因杂合变异类型有无义变异和错义变异。以上结果提示SCN1B基因杂合变异位点及变异类型与癫痫表型的轻重没有明确相关性,SCN1B基因基因型与表型的相关性尚需要大样本量的研究。
近年来关于SCN1B基因变异的功能研究逐渐增多,SCN1B基因表达的蛋白质β1B不仅是跨膜蛋白,还是在胚胎发育过程中主要表达的可溶性蛋白,可以促进神经突起的生长。Patino等发现的变异p.G257R导致β1B蛋白的细胞内滞留,导致蛋白功能缺失。随后Dang等[18]确定了R85位点的变异体存在致蛋白功能丧失的机制。大多数先前关于SCN1B基因变异体的功能研究发现其存在功能丧失,尤其干扰其增强钠通道开关的能力,如变异位点R125L可能因失去氢键而影响蛋白质的结构和稳定性。这些变异的功能研究结果表明,它们可能阻碍主要孔形成α亚单位的作用[30]。因此,使用钠通道阻滞剂治疗与SCN1B基因变异相关癫痫的疗效仍不确定。
SCN1B基因杂合变异多在GEFS+家系中被发现,为遗传性变异,其相关的癫痫发作起病年龄范围较大,可从婴儿期至学龄前期,多具有热敏感特点。发作类型多样,以GTCS、局灶性发作为主,临床表型常为热性惊厥、FS+。本研究首次报道了SCN1B基因杂合变异与婴儿肌阵挛性癫痫、婴儿癫痫性痉挛综合征和非特异性DEE表型相关。治疗可首选丙戊酸、左乙拉西坦等广谱ASM,多数患儿发作容易得到控制,预后较好。钠通道阻滞剂如奥卡西平、拉莫三嗪等可能加重部分患儿的发作,应用需谨慎。


















