当地时间 3 月30 日,美国 FDA 批准渤健和 Ionis 联合开发的诺西那生(Spinraza)高剂量(50 mg/28 mg)方案上市,用于治疗脊髓性肌萎缩症(SMA)。此前 FDA 已批准诺西那生 12 mg 治疗方案。2025 年 4 月,高剂量诺西那生治疗方案也已在中国报上市。

截图来源:渤健官网
诺西那生高剂量治疗方案包括一个更快速的负荷期,即间隔 14 天注射两次 50 mg 负荷剂量,之后每四个月注射一次 28 mg 维持剂量。从 12 mg 剂量过渡到高剂量的患者,将接受一次 50 mg 剂量代替下一次 12 mg 剂量,之后每四个月注射一次 28 mg 维持剂量。
本次高剂量方案的获批是基于 II/III 期 DEVOTE 研究的结果。DEVOTE 是一项 II/III 期随机、对照、剂量递增研究,在全球约 42 个中心纳入 145 名不同年龄、不同 SMA 分型的受试者,试验包含三个部分:A 部分,开放标签的安全性评估队列;B 部分,双盲活性对照随机治疗队列;C 部分,开放标签治疗队列。
其中,B 部分包含关键队列(未接受过治疗的婴儿型 SMA 患者,n=75)和支持性队列(未接受过治疗的晚发型 SMA 患者,n=24),主要终点为与 III 期 ENDEAR 研究中匹配的未经治疗假对照组相比,治疗 6 个月时费城儿童医院婴儿神经肌肉疾病测试(CHOP-INTEND)评分较基线的变化,其中ENDEAR 研究是 12 mg 诺西那生注射液获得监管批准的两项关键研究之一。
B 部分研究的关键队列结果显示,接受高剂量的诺西那生治疗的初治、有症状婴儿,与来自 ENDEAR 研究的预先设定的匹配安慰剂(未治疗)组相比,在由CHOP-INTEND 量表评估的运动能力方面得到显著改善(平均差异:26.19分;+15.1 vs. -11.1,p<0.0001)。
C 部分为开放标签评估,研究对象是从现有获批的 12 mg 方案转换至 50 mg/28 mg 给药方案的儿童和成人患者(n=40)。C 部分(n=40)初步结果显示:4-65 岁的参与者在接受获批的 12 mg 方案治疗中位时间 3.9 年后,过渡至高剂量方案(一次 50 mg 剂量,随后为 28 mg 维持剂量),运动功能得到改善,第 302 天时,HFMSE 评分较基线平均提高 1.8 分,RULM 平均提高 1.2 分。
2025 年 6 月,渤健进一步公布的 DEVOTE 研究 C 部分最新数据显示:大多数参与者在过渡至高剂量方案后,在 HFMSE、RUL 和 / 或 CGI-C 评分上均有改善,且不同表型、功能状态及年龄的患者中均观察到此类改善。例如,非行走患者的 HFMSE 平均改善为 + 2.5 分,行走患者为 + 1.1 分。这些新数据表明,即使是已接受多年治疗的患者,仍有可能通过调整剂量获得额外的功能提升。
在 DEVOTE 研究各部分中,高剂量方案总体耐受性良好,安全性特征与获批的 12 mg 方案相似。12 mg 方案最常见的不良事件(AEs)包括呼吸道感染、发热、便秘、头痛、呕吐及背痛;DEVOTE 研究中,各诺西那生治疗组的不良事件发生率相近。导致研究退出和死亡的 AE 仅在 B 部分初治队列中出现,50 mg/28 mg 组、12 mg 组和匹配的假治疗组发生率分别为 20%(10 例)、24%(6 例)和 55%(11 例)。
脊髓性肌萎缩症(SMA)是一类严重的罕见神经肌肉疾病,为常染色体隐性遗传病,病变始于中枢神经系统,因运动神经元变性导致患者出现进行性肌肉无力与萎缩。
诺西那生是一款以反义寡核苷酸(ASO)为核心机制的药物,通过鞘内注射给药,可直接作用于疾病起源部位 —— 中枢脊髓前角及延髓运动神经元,精准发挥治疗作用,从而改善患者运动功能、提高生存率,改变 SMA 的疾病进程。
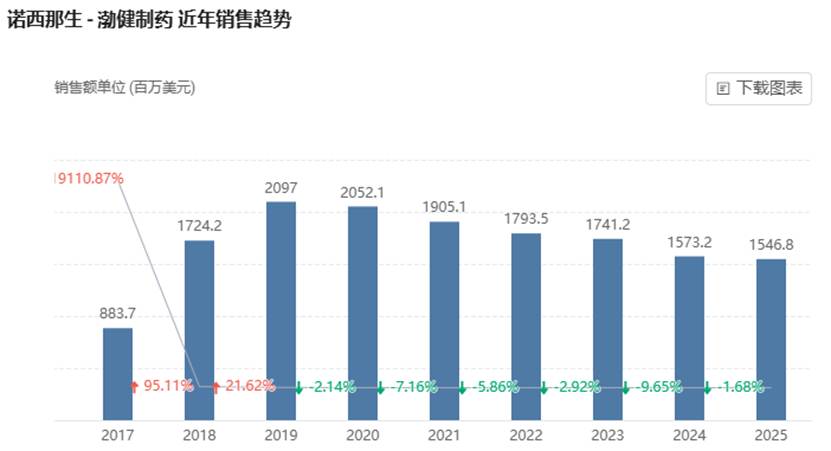
诺西那生于 2016 年 12 月首次获批,是全球首个获批的 SMA 治疗药物。目前,该药的 12 毫克剂量治疗方案已在全球超过 71 个国家/地区上市。诺西那生高剂量方案已在欧盟、美国获批上市,并在中国、日本申报上市。2025 年诺西那生的销售额为 15.46 亿美元,同比下降 1.68%。
截图来源:Insight 数据库


















