本文刊于:中华内科杂志,2026, 65(2): 142-155.
作者:中华医学会罕见病分会 北京医学会罕见病分会
通讯作者:袁云,北京大学第一医院神经内科 罕见病医学中心,北京 100034,Email: yuanyun2002@126.com
引用本文: 中华医学会罕见病分会, 北京医学会罕见病分会. Becker型肌营养不良诊治中国专家共识[J]. 中华内科杂志, 2026, 65(2): 142-155. DOI: 10.3760/cma.j.cn112138-20250730-00445.
Becker型肌营养不良(Becker muscular dystrophy,BMD)是由编码抗肌萎缩蛋白的DMD基因致病性变异所导致的一种X-连锁隐性遗传性肌病,包括肢带型肌无力、股四头肌肌病、孤立性痉挛疼痛综合征和无症状高肌酸激酶血症4个临床亚型,部分患者也可伴随心肺损害、心理障碍、关节及脊柱畸形,其诊断主要依靠基因检测和/或骨骼肌病理学检查。BMD的治疗涉及神经内科、心血管内科、呼吸内科、康复医学科、骨科、消化内科、麻醉科、临床营养科、心理科以及医学遗传科医师的综合管理干预,以维持患者的运动功能、骨与关节功能、心肺功能以及消化功能,保持良好的营养状态和心理健康。为规范我国BMD的诊治和管理,中华医学会罕见病分会和北京医学会罕见病分会联合成立了由多学科专家组成的BMD专家共识编写委员会,共同制定了我国BMD的诊断与治疗专家共识,旨在提高患者的生活质量和降低疾病负担。
Becker型肌营养不良(Becker muscular dystrophy,BMD)是由编码抗肌萎缩蛋白的DMD(Duchenne muscular dystrophy,DMD)基因致病性变异所导致的一种主要累及骨骼肌的X-连锁隐性遗传性疾病,与Duchenne型肌营养不良和X-连锁扩张型心肌病同属于抗肌萎缩蛋白病。BMD发病率在存活男婴中约为5.4/100 000 [ 1 ] ,人群患病率约为1.6/100 000 [ 2 ] 。约50%的BMD患者具有家族遗传史,通常由DMD基因的框内致病性变异或错义变异所致,框外变异或无义变异亦可导致该病的发生 [ 3 ] ,出现骨骼肌的抗肌萎缩蛋白表达量下降和/或分子量变化,但并不会导致抗肌萎缩蛋白的完全缺失,因此BMD是抗肌萎缩蛋白病相对良性的疾病亚型 [ 4 , 5 , 6 , 7 , 8 ] 。BMD具有高度的表型异质性,包括肢带型肌无力、股四头肌肌病、孤立性痉挛疼痛综合征和无症状高肌酸激酶(creatine kinase,CK)血症4个临床亚型 [ 4 , 5 , 6 , 8 , 9 , 10 ] ,该病可以伴随心脏受累,是导致患者死亡的最常见原因 [ 6 , 11 , 12 ] 。少数女性DMD基因致病性变异携带者也可出现BMD的临床表现,呈现出不同程度的骨骼肌和/或心肌受累 [ 13 , 14 , 15 , 16 ] 。
为规范BMD的诊治和管理,中华医学会罕见病分会和北京医学会罕见病分会联合成立了一个由多学科专家组成的BMD专家共识编写委员会,基于对BMD的文献分析和专家经验,制定了中国BMD的多学科诊断与治疗专家共识,指导我国临床医师对BMD的规范化诊疗,并为政府管理部门对该病的管理提供参考,以提高患者的生活质量、改善预后和降低疾病负担。
由于BMD的大规模队列研究和随机对照试验研究较少,其他类型的循证医学证据也相对缺乏,本共识的推荐建议制定并非完全基于传统的循证医学方法,主要参考改良德尔菲法进行制定,包括3轮推荐建议问卷调查。第1轮问卷调查中将专家赞同百分比≥70%的条目列入推荐建议。未达成共识的推荐建议根据专家反馈意见对问卷进行修改,对未达成共识的问题进行第2轮问卷调查,依然不能形成共识的推荐建议,进行第3阶段的专家面对面讨论。
第1阶段,BMD专家共识编写委员会委托神经病学专家起草本共识的初稿,初稿起草专家通过北京大学医学图书馆网站对中国知网、万方数据库、NCBI PubMed电子数据库的文献进行检索,文献检索时间为1990年至2024年。采用布尔检索表达式“A and B”进行检索,其中A的主要检索词包括Becker muscular dystrophy、dystrophinopathy、dystrophin、DMD、Becker型肌营养不良、抗肌萎缩蛋白病,B的主要检索词包括diagnosis、genetics、treatment、management、诊断、遗传学、治疗、管理。
第2阶段,由来自神经病学、心脏病学、呼吸病学和医学遗传学领域的专家组成共识核心工作小组,对共识的初稿进行讨论、修改和确认。
第3阶段,由神经内科医师、儿科医师、心血管内科医师、呼吸内科医师、骨科医师、消化内科医师、临床营养科医师、康复医学科医师、麻醉科医师、临床心理科医师以及医学遗传科医师组成的共识完善小组,对核心工作小组确认的共识进行评议、修改和再确认。
核心工作小组在中华医学会罕见病分会和北京医学会罕见病分会的指导下确认并发布该共识。所有专家组成员声明无利益冲突。本共识的制定由中央高水平医院临床研究专项、国家自然科学基金以及北京大学临床科学家培养计划联合资助。
二、BMD临床表现
(一)男性患者
1. 骨骼肌表现:BMD患者的骨骼肌受累具有高度的异质性,除无症状高CK血症和孤立性痉挛疼痛综合征之外,通常在5岁后出现肢体无力的表现,平均发病年龄在12岁左右 [ 5 , 8 , 9 ] 。表现为肢带型肌无力或股四头肌肌病患者的首发症状是骨盆带肌的无力,表现为上楼费力、蹲起费力、跟腱挛缩、行走鸭步、腰椎前凸,随着疾病进展,逐渐出现Gowers征,不能跑跳、蹲起以及独立上台阶 [ 5 , 9 ] 。肢带型肌无力的患者还存在肩胛带肌无力和萎缩,随着疾病进展,双上肢活动逐渐受限。表现为肢带型肌无力或股四头肌肌病的BMD患者可以出现肌肉痉挛疼痛,通常发生在运动期间或运动之后。少数BMD患者表现为孤立性痉挛疼痛综合征或单纯高CK血症 [ 4 , 6 , 8 , 17 ] ,但有可能随疾病进展而转化为肢带型肌无力或股四头肌肌病。各种BMD临床亚型都存在不同程度的腓肠肌肥大 [ 5 , 9 ] 。多数存在肢体无力的BMD患者的独立行走能力可以保持到40~50岁,少数患者可以持续到50岁之后 [ 3 , 4 , 5 , 6 , 7 , 8 ] ,症状严重者丧失独立行走能力的年龄不早于16岁,部分患者的预期寿命接近正常水平 [ 8 , 9 ] ,特别是表现为长期无症状高CK血症或孤立性痉挛疼痛综合征的患者。
推荐意见1:BMD可分为肢带型肌无力、股四头肌肌病、孤立性痉挛疼痛综合征、无症状高肌酸激酶血症4个亚型。
推荐意见2:5岁后出现肢体近端无力、腓肠肌肥大和血清肌酸激酶水平升高,为表现为肢带型肌无力和股四头肌肌病的BMD红旗征。
推荐意见3:对任何BMD临床亚型都需要进行长期随访,明确骨骼肌和骨骼肌外损害发生的时间规律,特别需注意较轻类型向严重类型的转化。
2. 骨骼肌外表现:多数BMD患者的心脏受累晚于肢体无力,也可以早于肢体无力。BMD患者的心脏受累早期主要表现为左室下壁及侧壁为主的心肌纤维化 [ 18 ] ,部分患者出现以左心室收缩功能损害为特征的收缩功能减低性非扩张型心肌病,30%~40%的BMD患者可进展至扩张型心肌病,表现为左心室射血分数(left ventricular ejection fraction,LVEF)<45%和心室扩张,20岁前该比例为15%,40岁后为55% [ 9 , 12 , 18 , 19 , 20 ] 。心脏受累可进展至终末期心力衰竭,是患者死亡的主要原因。少数患者可出现室性期前收缩或不同程度的心脏传导阻滞,但很少出现持续性室性心动过速、完全性房室传导阻滞和心源性猝死。呼吸功能障碍多出现在疾病的中晚期,早期表现为睡眠中易醒和仰卧位呼吸困难,随疾病进展出现咳嗽无力、呼吸困难、肺炎频发。消化系统症状早期可出现便秘以及体脂肪和/或体重超标,随疾病进展出现咀嚼与吞咽功能下降以及食欲下降,导致体重下降和营养不良。疾病早期出现踝关节挛缩,随疾病进展出现膝关节挛缩、肘关节挛缩、脊柱侧弯、指间关节和腕关节挛缩 [ 5 , 9 ] 。约8%的BMD患者伴有智力发育障碍 [ 21 ] 。可出现孤独症谱系障碍、注意力缺陷多动障碍、抑郁或焦虑障碍以及强迫症。癫痫患病率高于普通人群 [ 22 , 23 ] 。
推荐意见4:BMD的骨骼肌外表现包括心肺受累、关节挛缩、脊柱畸形、消化功能障碍、营养失衡、神经心理异常。
推荐意见5:BMD患者的关节挛缩和肢体无力存在相关性;呼吸功能障碍一般出现在上肢无力之后;心理障碍以抑郁为主。
(二)女性患者
少数(2.5%~25.7%)女性DMD基因致病性变异携带者也可出现不同程度的骨骼肌和/或心肌受累 [ 13 , 16 ] ,以BMD样表型最为常见,骨骼肌受累多具有不对称性。少数患者表现为扩张型心肌病或Duchenne型肌营养不良样表型 [ 13 , 16 , 24 ] 。
推荐意见6:女性BMD患者的病情发展规律与男性患者类似,也需注意骨骼肌外表现。
三、辅助检查
对于疑诊BMD的患者,在确定病因和骨骼肌损害严重程度的同时,也需确定是否存在骨骼肌外损害以及受累程度。
1. 血清生化检测:BMD患者的血清CK水平高于正常范围 [ 5 , 8 , 9 ] ,一般高于1 000 IU/L,无症状高CK血症和孤立性痉挛疼痛综合征患者的血清CK水平可以低于1 000 IU/L,血清CK水平正常者十分罕见 [ 6 ] ,病情严重者血清CK水平也可超过10 000 IU/L [ 25 ] ,在急性感染后患者血清CK水平在短期内可因骨骼肌溶解而明显升高。血清CK水平的升高在疾病早期时更为明显,疾病后期出现降低 [ 6 ] 。可伴随出现丙氨酸转氨酶和天冬氨酸转氨酶水平的升高。BMD患者首次确诊时应同时进行血清肌钙蛋白、B型利钠肽或N末端B型利钠肽原的检测,也应进行一次血清钙、磷、镁、维生素D 3、碱性磷酸酶以及甲状旁腺激素的基线测定。疾病晚期需进行血常规和生化全项检测。
推荐意见7:血清CK检测是BMD患者的首选辅助检查,每次复诊时进行血清CK检测,血清CK检测应当在没有感染和过度活动的情况下进行。
推荐意见8:BMD患者需注意预防骨骼肌溶解。
推荐意见9:疾病后期血清CK水平的下降与残留肌纤维减少有关,而肌力正常和骨骼肌磁共振检查没有改变的情况下出现CK下降,并非疾病恶化表现。
推荐意见10:首次确诊时即需进行心脏和骨代谢检查。
2. 电生理检查:BMD患者针极肌电图的改变为非特异性的肌源性损害,在疾病的早期可见运动单位电位波幅降低且时限缩短,在疾病的晚期可表现为电静息状态,由于肌纤维数量的明显减少,可出现混合性甚至是假性神经源性损害的特点 [ 26 ] 。BMD患者首次确诊时应进行心电图检查,对于心室收缩功能异常(LVEF<50%)或心电图提示心脏传导异常时,应进行24 h动态心电图检查 [ 27 ] 。
推荐意见11:当血清CK水平超过正常上限10倍以上时,针极肌电图和神经传导检查不是BMD的必要性检查。
推荐意见12:青少年BMD患者在临床首诊时需与脊髓性肌萎缩症相鉴别,肌电图检查具有重要的鉴别诊断价值。
推荐意见13:单纯高CK血症或痉挛疼痛综合征的患者,建议进行肌电图检查。
推荐意见14:每次复诊时进行心电图检查,必要时进行24 h动态心电图检查。
3. 影像学检查:骨骼肌磁共振检查可以确定受累肌肉的分布模式和严重程度,以此协助临床分型。无症状高CK血症和孤立性痉挛疼痛综合征的患者无明显骨骼肌脂肪浸润改变。表现为肢体无力的BMD患者骨骼肌磁共振改变主要为肌肉组织的脂肪浸润,也可伴有不同程度的水肿改变 [ 28 ] 。疾病早中期主要是骨盆带肌和大腿肌肉出现脂肪浸润( 图1A~1B ),主要累及臀大肌、臀中肌、耻骨肌、股四头肌、长收肌、大收肌、半膜肌和股二头肌 [ 7 ] ,而股薄肌、长收肌、缝匠肌和半腱肌可相对保留和/或肥大( 图1B ) [ 7 , 28 , 29 ] 。随疾病进展,小腿的腓肠肌、比目鱼肌和胫前肌出现脂肪浸润( 图1C ),上肢中的肱三头肌和肱二头肌也可出现脂肪浸润,但三角肌和喙肱肌可相对保留 [ 7 , 9 , 28 ] 。
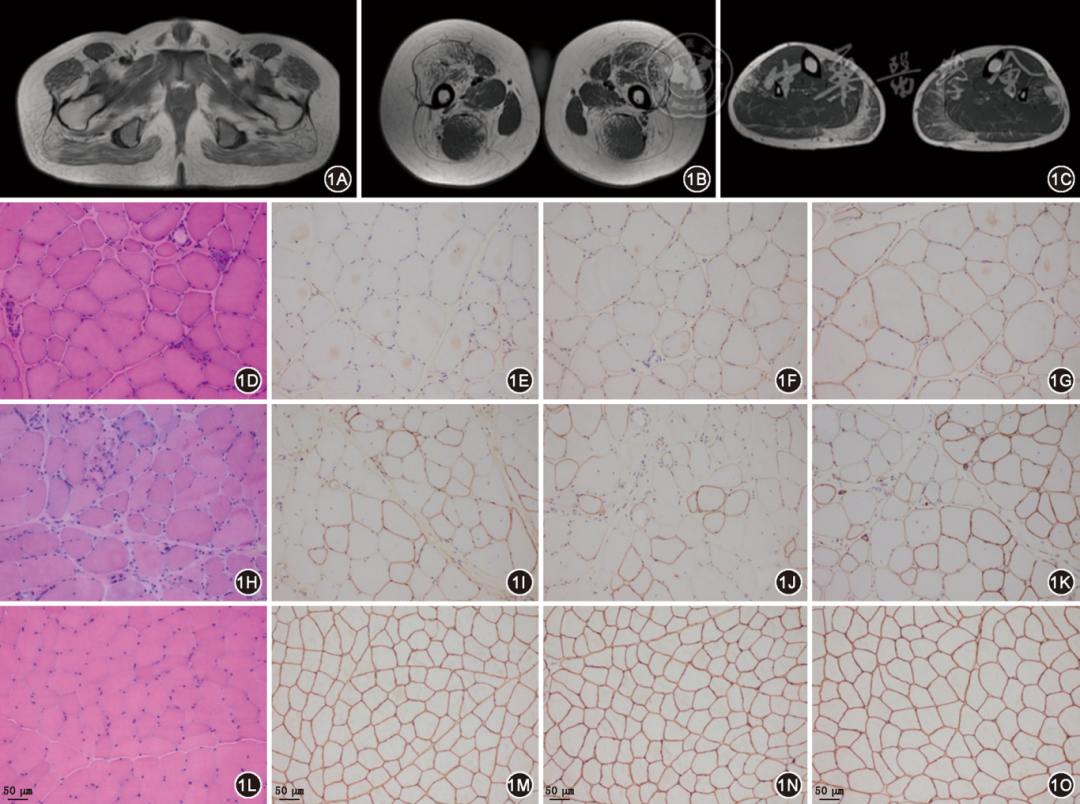
图1 Becker型肌营养不良患者的骨骼肌影像学及病理学特征 1A~1G为Becker型肌营养不良患者;1H~1K为女性症状性DMD基因致病性变异携带者;1L~1O为正常对照;1A~1C均为患者卧位T1加权成像骨骼肌磁共振图片,1A显示臀大肌的脂肪浸润,1B显示股薄肌、长收肌、缝匠肌和半腱肌的相对保留,1C显示腓肠肌内侧头的脂肪浸润;1D、1H和1L为苏木精-伊红染色 ×200,1D和1H显示肌营养不良样病理改变,1L为正常肌肉组织;1E、1I和1M为抗肌萎缩蛋白-N端免疫组织化学染色 ×200,1F、1J和1N为抗肌萎缩蛋白-C端免疫组织化学染色 ×200,1G、1K和1O为抗肌萎缩蛋白-R端免疫组织化学染色 ×200,1E~1G和1I~1K显示抗肌萎缩蛋白的不同程度表达下降;1M~1O显示抗肌萎缩蛋白的正常表达
初诊患者需进行超声心动图的检查,依据检查结果决定是否进行斑点追踪超声心动图和心脏磁共振的检查。心脏磁共振检查相较于超声心动图对于心肌受累具有更高的敏感性,超声心动图正常的BMD患者也可能存在心肌纤维化 [ 19 , 27 ] 。
推荐意见15:骨骼肌磁共振检查对BMD的临床诊断和病情评估具有提示意义,但不需要作为常规检查。
推荐意见16:骨骼肌磁共振检查部位主要是骨盆和大腿,不常规进行其他部位的检查。
推荐意见17:心脏影像学检查时间一般不早于7岁,心脏影像学检查以超声心动图为主,心脏磁共振检查不作为常规检查。
4. 基因检测:BMD患者的基因检测流程( 图2 )详见《抗肌萎缩蛋白病中国诊断指南》 [ 3 ] ,基因检测发现的DMD基因变异需要根据美国医学遗传学与基因组学学会指南 [ 30 ] 进行变异致病性的评价。
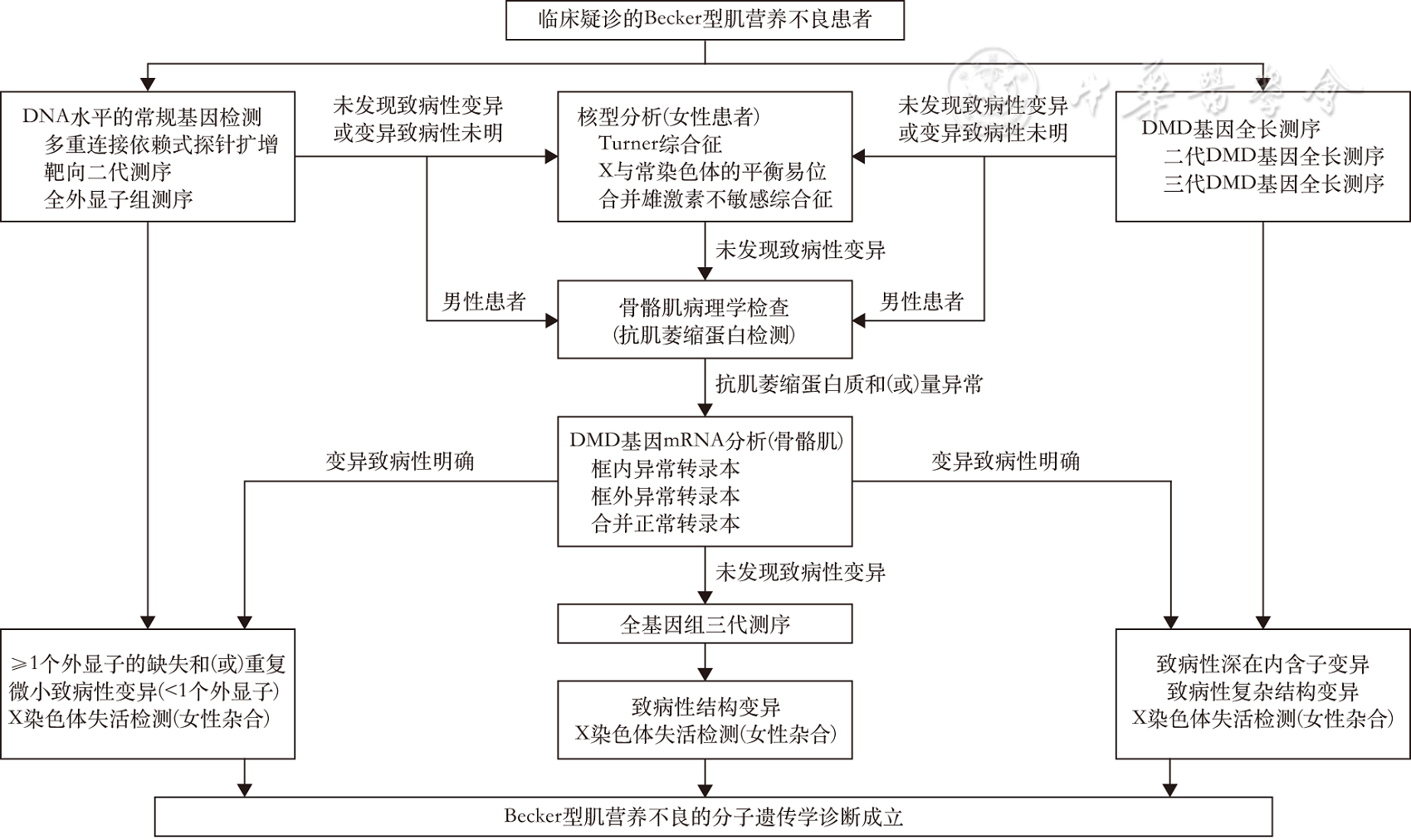 图2 Becker型肌营养不良患者的基因检测流程图
图2 Becker型肌营养不良患者的基因检测流程图
(1)男性患者
①常规基因检测:主要用于鉴定DMD基因的经典变异(外显子及其邻近内含子区域中的变异) [ 7 , 31 , 32 , 33 ] ,包括多重连接依赖式探针扩增技术、靶向神经肌肉病的二代测序或全外显子组测序。当未检测出DMD基因的致病性外显子缺失和/或重复变异以及微小致病性变异时,或检测出的DMD基因变异致病性未明时(特别是错义变异、同义变异以及位于非经典剪接区域的微小变异),需进行下一步的基因检测分析。
②DMD基因全长测序:DMD基因全长测序既可检测经典变异,也可检测深在内含子变异和复杂结构变异等非经典变异。DMD基因二代全长测序 [ 34 ] 和DMD基因三代全长测序 [ 34 , 35 ] 均可用于检测DMD基因的深在内含子变异以及结构变异,但对于复杂结构变异,DMD基因三代全长测序更具优势。当未检测出致病性深在内含子变异以及结构变异时,或检测出的深在内含子变异以及结构变异致病性未明时,需进行下一步的基因检测分析。
③抗肌萎缩蛋白及DMD基因mRNA分析:当骨骼肌病理学检查发现抗肌萎缩蛋白存在质和/或量的异常,但DMD基因致病性变异未明时,需进行肌肉组织的DMD基因mRNA分析,可采用转录组测序 [ 7 , 35 ] 或逆转录-聚合酶链式反应交叉片段扩增法 [ 34 ] 进行分析。当发现DMD基因存在异常转录本时,并且常规基因检测和DMD基因全长测序中发现的变异能够解释该异常转录本时 [ 7 , 34 , 35 , 36 ] ,则可以确定该变异是致病的,否则需要进行下一步的基因检测分析。
④全基因组三代测序:用于鉴定超大范围的复杂结构变异以及断裂点位于基因组高度重复序列中的复杂结构变异 [ 31 ] 。
(2)女性患者:首先进行DNA水平的常规基因检测 [ 37 ] ,如发现女性患者携带DMD基因的杂合致病性变异时,需进一步进行X染色体失活检测;如发现患者DMD基因存在致病性复合杂合或纯合变异时,则可不进行X染色体失活检测。常规基因检测未能鉴定出致病性变异时,首先考虑进行核型分析,以除外一些特殊变异类型,包括Turner综合征(45,X) [ 38 ] 、常染色体与X染色体的平衡易位 [ 39 ] 、雄激素不敏感综合征合并DMD基因致病性变异(46,XY;男性假两性畸形) [ 40 ] 。如常规基因检测和核型分析均未鉴定出致病性变异,考虑依次进行上述男性患者的基因检测流程。个别生殖细胞嵌合体变异的女性携带者外周血DNA可以不存在DMD基因致病性变异。
推荐意见18:基因检测应当作为确诊BMD的首选辅助检查,也是遗传咨询和/或产前诊断的前提。
推荐意见19:阅读框理论适用于大多数BMD患者的表型预测,但无法进行精确的临床亚型判断。
推荐意见20:女性患者的X染色体失活检测需结合患者的基因型确定。
5. 骨骼肌病理学检查:存在肢体无力的BMD患者骨骼肌组织学改变通常为肌营养不良样病理改变,表现为肌纤维直径变异加大、成组的肌纤维坏死和再生以及间质结缔组织的增生,也可出现嗜酸高收缩肌纤维 [ 41 ] 。孤立性痉挛疼痛综合征和无症状高CK血症患者可无或仅出现轻微肌病样病理改变。采用针对抗肌萎缩蛋白-N端、R端以及C端结构域的单克隆抗体进行免疫组织化学或免疫荧光染色,可以发现患者存在不同程度的抗肌萎缩蛋白表达下降 [ 3 , 7 , 37 ] ( 图1E~1G )。如患者临床表型高度怀疑BMD,但抗肌萎缩蛋白-N端、R端及C端均正常或几乎正常表达时,特别是无症状高CK血症或痉挛疼痛综合征患者,应当采用蛋白质印迹法分析抗肌萎缩蛋白分子量的变化 [ 42 ] 以协助诊断。
女性DMD基因致病性变异携带者肌纤维出现抗肌萎缩蛋白表达异常的比例在10%~100%之间,主要取决于X染色体偏倚失活的程度。症状性女性DMD基因致病性变异携带者组织学的经典改变为肌营养不良样病理改变,抗肌萎缩蛋白的表达通常呈现“马赛克样”分布( 图1I~1K ),即阴性表达或表达下降的肌纤维与正常表达的肌纤维呈相间分布 [ 16 ] 。无症状携带者可无明显病理改变。
推荐意见21:骨骼肌病理学检查在基因检测明确诊断的患者中不作为常规检查。
推荐意见22:在临床分型难以确定和DMD基因致病性变异未明的情况下需要进行骨骼肌病理学检查。
四、诊断流程
(一)疑诊标准
当患者出现以下表现之一时,应怀疑BMD的可能 [ 3 ] ( 图3 ),包括:(1)5岁后出现肢体无力(近端>远端),伴或不伴腓肠肌肥大;(2)运动诱发的肌痉挛和/或肌痛;(3)出现胸闷、气促、下肢水肿、心慌,伴肢体无力;(4)抗肌萎缩蛋白病家族史;(5)无法用其他原因解释的血清CK水平升高。对于疑诊BMD的患者,进行基因检测和/或骨骼肌病理学检查,以明确患者的诊断。
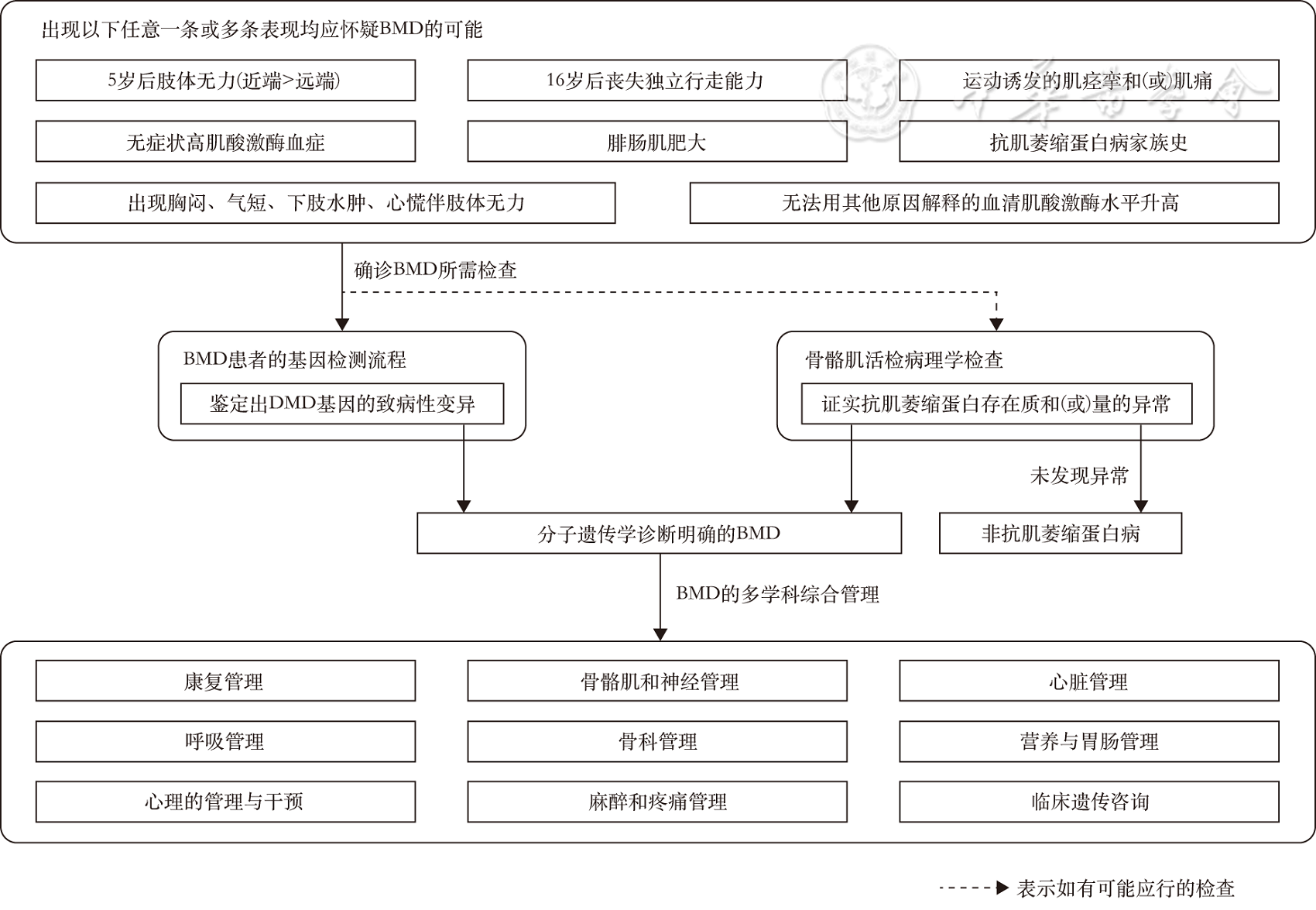 图3 Becker型肌营养不良的诊断与多学科管理
图3 Becker型肌营养不良的诊断与多学科管理
(二)确诊标准
BMD的确诊标准:(1)5岁后出现肢体无力和/或16岁之后丧失独立行走能力;(2)存在肌肉痉挛疼痛和/或高CK血症而无肢体无力;(3)基因检测鉴定出DMD基因的致病性变异;(4)骨骼肌病理学检查证实抗肌萎缩蛋白存在量和/或质的异常,同时除外其他导致抗肌萎缩蛋白继发性表达缺陷的肌病 [ 3 ] 。疑诊患者出现(1)和/或(2),同时存在(3)和/或(4)均可确诊为BMD。
推荐意见23:临床分型基于临床表现,不能仅依靠辅助检查结果进行临床分型。
五、多学科综合管理
首先对BMD患者进行多系统的评估,明确患者骨骼肌和其他系统的受累程度,制定个体化的多学科管理方案,多学科团队成员包括神经内科、儿科、心血管内科、呼吸内科、康复医学科、骨科、消化内科、麻醉科、临床营养科、心理科以及医学遗传科医师,依据患者的临床表现和疾病发展阶段确定具体哪些学科专家参与。对患者进行综合管理干预( 图3 ),维持患者的运动功能、骨与关节功能、心肺功能以及消化功能,保持良好的营养状态和心理健康,以延长患者独立行走时间和改善生活质量。可采用儿童生活质量问卷 [ 43 ] 、生活满意度指数或生活质量调查问卷 [ 44 ] 评估患者生活质量。随访内容及评估频率需依据患者的病情变化及时调整。
推荐意见24:各省及自治区至少应有一家医院建立神经肌肉病中心。
推荐意见25:神经肌肉病中心医师需要在能够开展神经肌肉病相关工作的医院进行培训。
推荐意见26:BMD的管理应当是多学科协作诊疗模式。独立行走阶段每1~2年进行1次多系统评估,不能独走阶段每6~12个月进行1次多系统评估。
(一)疫苗、饮食和活动
BMD患者接种疫苗无禁忌证。对于合并心肺受累的患者,除需要接种肺炎球菌疫苗,也需要按照接种时间表每年接种流感疫苗 [ 45 ] 。体重和体脂肪正常的患者,重视钙离子和维生素D的补充,预防骨质疏松。应避免肌肉离心收缩训练和高强度抗阻力运动 [ 46 ] ,避免剧烈的竞技类体育运动和过度锻炼。即使在不能独走阶段,也应当主动和被动地活动肢体,预防废用性肌萎缩。
推荐意见27:出现肢体无力和骨骼肌外损害的患者,接种肺炎球菌疫苗,并每年接种流感疫苗。
推荐意见28:疾病早期要预防超重,特别是进行糖皮质激素治疗的患者。
推荐意见29:在疾病的任何阶段都需要适度活动。
(二)康复管理
康复评估项目包括关节活动范围、肌力、姿势、步态、运动功能状态、日常生活活动能力以及生活质量评定 [ 9 , 47 ] 。
康复治疗主要包括关节活动范围训练、适度肌力和有氧运动训练、姿势管理、心肺和吞咽功能康复。关节活动范围训练采用规律性的被动和主动牵伸技术对即将或已出现挛缩的软组织进行牵伸治疗。根据患者情况适当进行低至中等强度的向心或等长收缩,避免高强度收缩和离心收缩,限制受累严重骨骼肌的剧烈收缩 [ 48 ] 。低至中等强度的有氧运动有助于患者维持活动耐力和心肺功能。每周进行3~5次康复治疗,每次30~45 min,时间长短以不感到疲劳和疼痛为标准,不导致血清CK水平的增加。
踝关节活动范围受限的患者,夜间可穿戴踝-足支具将踝关节维持在中立位。静态手夹板适用于手指屈肌挛缩的患者,夜间使用腕-手夹板有助于将腕关节和指间关节维持在功能位。对于逐渐丧失步行能力的患者,可尝试使用膝-踝-足支具来辅助膝关节的伸展稳定性,也可用电驱动式站立架替代,同时使用助行器辅助迈步。对于不能独走阶段的患者应提供安全的生活环境、个性化的座椅、电动轮椅、交流技术设备等,以帮助患者尽可能独立地完成简单日常活动 [ 9 ] 。
推荐意见30:初诊患者应当开始康复管理,出现肢体无力的患者都需要常规进行康复治疗。
推荐意见31:康复管理不仅针对骨骼肌、关节、步态和姿势,还需要针对呼吸、心脏以及吞咽功能损害进行相关康复治疗和指导。
推荐意见32:康复治疗的场所以家庭和社区为主。
(三)骨骼肌和神经管理
初诊患者针对骨骼肌进行评估:(1)采用徒手肌力评估或定量肌力测试评估肌力 [ 49 ] 。(2)独立行走阶段可采用肢带型肌营养不良的北极星评估 [ 50 ] 、北极星移动评价量表和Vignos下肢功能状态量表 [ 47 ] 评估患者的运动功能,采用6分钟步行试验评估患者的步行能力和耐力 [ 49 ] ,采用计时功能试验评估患者的短时运动功能(包括跑/走10米测试、卧-立位测试、4台阶测试)。不能独走阶段采用神经肌肉病运动功能评定量表和Brooke上肢功能状态量表评估患者的运动功能 [ 47 ] 。
依据患者的疾病严重程度确定是否进行糖皮质激素治疗以及治疗的开始时间、剂量和方案。无症状高CK血症和孤立性痉挛疼痛综合征患者无需糖皮质激素治疗,但需长期随访注意较轻类型向严重类型的转化,亦可使用艾地苯醌或辅酶Q 10缓解肌肉痉挛疼痛的症状。早期出现肢体无力伴血清CK水平高于10 000 IU/L的患者可以参考Duchenne型肌营养不良患者 [ 47 ] 进行适量糖皮质激素治疗,包括泼尼松和地夫可特 [ 51 , 52 , 53 , 54 ] 。糖皮质激素治疗的剂量及疗程需依据患者的临床表现、CK水平、骨骼肌磁共振改变和/或抗肌萎缩蛋白表达量等综合确定。伐莫洛龙是一种新型解离型皮质类固醇激素 [ 55 ] ,用于治疗Duchenne型肌营养不良患者 [ 56 ] ,对于BMD患者具有潜在治疗价值。BMD患者的癫痫治疗遵循现有的癫痫诊疗指南。
推荐意见33:症状严重的BMD患者可以参考Duchenne型肌营养不良患者进行糖皮质激素治疗。
推荐意见34:无感染或过度运动等情况下的血清CK水平超过10 000 IU/L,需进行糖皮质激素治疗。
推荐意见35:出现心脏受累也可进行糖皮质激素治疗。
(四)心脏管理
7岁以上患者每次复诊时应进行心电图和超声心动图检查。对于10岁以上的患者,每3~5年考虑进行1次心脏磁共振检查,以早期发现心肌纤维化。对于LVEF<50%的患者,每6~12个月进行1次血清肌钙蛋白、B型利钠肽或N末端B型利钠肽原、心电图、动态心电图、超声心动图检查,必要时进行斑点追踪超声心动图和心脏磁共振检查。
抗心力衰竭治疗是针对BMD患者心脏受累的主要治疗,可以改善患者的预后 [ 12 ] 。当LVEF下降至50%以下或心脏磁共振检查显示心肌纤维化时,可启动血管紧张素转换酶抑制剂治疗 [ 12 , 57 ] 。射血分数降低的心力衰竭患者应遵循现有心衰指南,选择性使用血管紧张素转换酶抑制剂或血管紧张素Ⅱ受体拮抗剂(不能耐受血管紧张素转换酶抑制剂患者)、β受体阻滞剂、盐皮质激素受体拮抗剂、利尿剂等药物,必要时进行心脏再同步化治疗、植入式心律转复除颤器(LVEF<30%) [ 12 , 27 ] 、左心室辅助装置或心脏移植治疗。
推荐意见36:7岁以上初诊患者应当进行心脏评估,依据评估结果决定后续心脏检查项目和随访频率。
推荐意见37:左心室辅助装置和心脏移植可用于肢体无力不明显的终末期心力衰竭患者。
(五)呼吸管理
出现肢体无力的患者开始进行肺功能检查 [ 47 ] ,包括用力肺活量(forced vital capacity,FVC)、第1秒钟用力呼气容积、最大呼气流量、咳嗽峰值流速、最大吸气压/最大呼气压。肺功能检查异常时,需测定脉搏血氧饱和度、血氧分压和二氧化碳分压,必要时进行膈肌超声检查。当患者出现睡眠呼吸障碍时,应考虑行多导睡眠监测,同时考虑监测经皮二氧化碳分压或呼气末二氧化碳分压。
在流感季节,流感抗原阳性者48 h内使用神经氨酸酶抑制剂或RNA聚合酶抑制剂(5岁以上)抗流感病毒治疗,如密切接触者确诊流感,也可预防性使用神经氨酸酶抑制剂。当咳嗽明显无力、FVC<预计值的50%、咳嗽峰值流速<270 L/min或最大呼气压<60 cmH 2O时(1 cmH 2O=0.098 kPa),需进行呼吸运动训练以及辅助咳痰处理 [ 58 ] 。当BMD患者夜间经皮二氧化碳分压>50 mmHg(1 mmHg=0.133 kPa)、FVC<预计值的50%、最大吸气压<60 cmH 2O时,需进行夜间无创辅助通气治疗 [ 47 , 59 ] ;当日间脉搏血氧饱和度<95%、二氧化碳分压>45 mmHg或清醒时存在呼吸困难,可进行日间无创通气 [ 60 ] 。当存在严重呼吸功能衰竭、咳嗽无力不能排痰,应考虑行有创性机械通气和气管切开术。
推荐意见38:呼吸功能测定在患者出现肢体无力之后进行。
(六)骨科管理
依据患者的年龄和发育状态进行关节活动范围、脊柱查体、骨龄、骨密度以及脊柱影像学检查 [ 47 ] 。当患者进入不能独走阶段后进行1次骨科脊柱查体和脊柱影像学检查,评定脊柱弯曲度的基线,不使用和使用糖皮质激素的患者分别每2~3年或1~2年拍摄一次直立位的脊柱正侧位全长X线片。如果患者出现腰背痛和/或骨折,每次随访时都应进行脊柱正侧位X线片的检查。
当患者出现非创伤性长骨或椎体骨折时,在补充钙和维生素D之后可静脉注射双膦酸盐治疗,并进行手术治疗。较长时间使用双膦酸盐可能诱发骨坏死。当患者脊柱弯曲度超过20°时,应由脊柱外科医师接诊和管理,使用脊柱支具。如果脊柱弯曲度大于30°并且持续进展影响心肺功能时,可考虑脊柱矫形手术治疗。如果伴随椎骨压缩性骨折,可考虑支具或手术治疗。在独立行走阶段,如果患者踝关节严重挛缩,膝关节和髋关节伸肌肌力保持良好,可考虑行足部和跟腱的外科手术治疗,以改善患者的姿势稳定和行走步态,延长独立行走时间 [ 51 ] 。术前应由神经内科、心血管内科、呼吸科、骨科以及麻醉科专家共同评估手术的风险和获益。术后需加强康复管理。
推荐意见39:骨质疏松的防治推荐碳酸钙、维生素D、阿仑膦酸钠和双膦酸盐。
推荐意见40:BMD患者踝关节严重挛缩,膝关节和髋关节伸肌肌力保持良好,可考虑行外科手术治疗。
(七)营养与胃肠管理
评估身高、体重、身体质量指数、人体成分分析、血常规以及生化全项,出现上肢无力的患者需要评估咀嚼与吞咽功能 [ 61 ] ,明确吞咽困难的临床表现、进餐时间,视频透视观察吞咽功能的咽部阶段。吞咽功能存在异常者需进一步评估是否存在胃食管反流和胃动力障碍,必要时进行胃镜及肠镜检查。
独立行走阶段患者应遵循《中国居民膳食指南(2022)》的要求,保证营养均衡。不能独走阶段患者超重/肥胖的风险升高,每年进行一次人体成分分析检测。当患者存在体脂肪和/或体重超标时,使用限能量高蛋白饮食,限制碳水化合物的摄入(40%~50%供能比),控制脂肪摄入量(20%~25%供能比),严格限制饱和脂肪酸和反式脂肪酸的摄入,增加优质蛋白质摄入 [ 62 ] 。存在胃食管反流时可以使用组胺H 2受体拮抗剂或质子泵抑制剂抑制胃酸分泌,使用硫糖铝和中和抗酸剂作为辅助疗法。胃轻瘫时需少量多餐和清淡饮食,使用促动力药物。存在便秘时应增加饮用水量和富含膳食纤维的饮食,进行排便训练,而后考虑使用膳食纤维补充剂、益生菌或渗透性泻药;急性便秘需使用渗透性泻药或大便软化剂,有时需要使用灌肠治疗。
咀嚼和吞咽功能障碍以及食欲下降患者,使用成人或儿童的营养风险筛查量表进行筛查,进行综合营养评价。营养治疗应遵循“天然食物-口服营养补充-肠内营养-肠外营养”的阶梯式治疗方案。短期内体重下降超过10%,或与年龄相关的体重增加未达标,需进行咀嚼与吞咽功能检查,并依据检查结果调整饮食习惯、饮食结构、食物性状与成分。进行咀嚼肌和吞咽肌训练症状无改善者及时考虑留置鼻胃管;如果预计鼻胃管营养支持时间大于4周,或不能耐受鼻胃管的患者,需考虑进行胃造瘘术 [ 9 ] 。
推荐意见41:初诊患者常规进行营养状态和消化功能评估。
(八)心理的管理与干预
初次诊断时在可配合情况下应进行心理测评,包括认知功能、智力、语言、社会功能、学习能力、情绪适应、行为调节以及社会活动参与度等,测评有问题者进行智力量表、孤独症和注意缺陷-多动障碍评估表的筛查。成年患者评估可采用患者健康问卷-9项抑郁量表 [ 63 ] 和广泛性焦虑障碍-7项量表 [ 64 ] 。
当患者存在认知方面的问题时,应进行心理康复治疗,特别是语言和认知功能的康复,鼓励患者参与社会活动。关注患者家庭成员的心理状况,由心理医师为患者及其家庭成员提供心理咨询和生活指导。患者或家庭成员出现焦虑、抑郁、强迫等心理问题会影响患者的治疗依从性,在心理康复无效的情况下,存在中至重度精神症状的患者应考虑精神药理学治疗。
推荐意见42:BMD患者在初次诊断时进行心理测评和神经心理评估。
推荐意见43:对于存在中至重度精神症状的患者,应进行精神药理学治疗。
(九)麻醉和疼痛管理
BMD患者围手术期的恶性高热 [ 65 ] 风险与其他神经肌肉病相同,在全身麻醉期间应避免使用卤族类吸入麻醉药,术中推荐使用全凭静脉麻醉进行麻醉维持 [ 66 , 67 ] 。避免使用去极化肌松剂和肉毒杆菌毒素,降低患者全身麻醉期间发生横纹肌溶解和高钾血症的风险 [ 65 ] 。部分BMD患者存在气道插管困难,需选择合适的气道管理工具进行气管插管。对于合并呼吸功能减退或心力衰竭患者,应在麻醉前充分评估手术的获益与风险。
患者独坐时选择合适的软垫和良好的坐姿,避免压迫坐骨神经造成疼痛。患者的疼痛管理应遵循三阶梯止痛原则,避免长期止痛治疗。使用止痛药物时需考虑对心脏和呼吸系统的副作用。使用阿片类药物时需依据患者的疗效反应调整到最低有效剂量,同时应避免出现或加重夜间低通气或呼吸暂停。
推荐意见44:建议避免使用卤族类吸入麻醉药、去极化肌松剂和注射肉毒杆菌毒素。
推荐意见45:止痛治疗应注意对骨骼肌和心脏的副作用。
(十)女性DMD基因致病性变异携带者的管理
年龄相对较大的无症状女性DMD基因致病性变异携带者首次确诊后进行骨骼肌和心脏评估 [ 68 ] 。骨骼肌和心脏受累女性患者的处理与男性患者一致。当女性患者表现为BMD的各个临床亚型时,按照男性BMD进行多学科的综合管理干预。
女性DMD基因致病性变异携带者在妊娠前需要进行遗传咨询和心脏评估,警惕妊娠期间和产后容量变化导致症状性女性携带者的心脏损害风险增加,目前有关女性携带者妊娠和分娩的文献有限 [ 24 ] 。自然分娩不是无症状女性携带者的禁忌,在进行无痛分娩时采取硬膜外麻醉比全身麻醉风险小,尤其是呼吸抑制方面 [ 9 ] 。
推荐意见46:症状性女性DMD基因致病性变异携带者按照男性BMD患者进行多学科的管理干预。
推荐意见47:女性DMD基因致病性变异携带者无论是否有症状,均应在妊娠早期进行心内科、呼吸科、麻醉科、神经内科以及产科的多学科评估。
(十一)临床遗传咨询
主要目的是降低该病继续遗传的风险,应在患者及家属充分知情的情况下,由患者及家属做出决定 [ 3 , 33 ] 。该病为X-连锁隐性遗传,患者DMD基因的致病性变异多数遗传自母亲,少数为新生变异。女性DMD基因致病性变异携带者有50%的概率将致病性变异传递给子代胎儿,携带致病性变异的男性胎儿成为患者,多数携带致病性变异的女性胎儿成为新的无症状携带者,少数携带致病性变异的女性胎儿因X染色体偏倚失活等机制出现骨骼肌或心脏受累成为症状性携带者 [ 24 ] 。表现为孤立性痉挛疼痛综合征或无症状高CK血症的年长男性先症者,因症状轻微对生活无明显影响,家族内女性携带者再次妊娠时需慎重斟酌产前诊断的必要性。
通常在妊娠11~13 +6周取胎盘绒毛或18~24周取羊水进行产前诊断,明确胎儿是否携带有与先证者相同的DMD基因致病性变异 [ 3 , 37 , 47 ] 。男性BMD患者有可能生育后代,男性胎儿通常为健康人,但女性胎儿必定是DMD基因致病性变异携带者,其女儿在妊娠前需要进行临床遗传咨询。可以考虑对BMD患者家属进行临床遗传咨询和症状前遗传学诊断。
推荐意见48:已生育过一例BMD患者或女性DMD基因致病性变异携带者的母亲,无论是否为DMD基因致病性变异携带者,再次妊娠后均建议进行产前或植入前诊断。
推荐意见49:高风险人群孕前及孕早期检查中建议进行DMD基因检测。
六、小结
本诊疗专家共识基于BMD现有的临床证据和研究进展,并广泛征求多学科专家意见,结合我国的国情,对BMD的临床表现、辅助检查、诊断流程和多学科协作诊疗模式进行了系统性阐述,特别是对女性DMD基因致病性变异携带者的管理和临床遗传咨询提出编写专家组的观点,为我国各级医疗机构神经肌肉病从业人员的临床工作提供参考。BMD患者的综合管理涉及多学科专家的诊疗合作,本共识代表编写专家组的观点,有助于政府卫生管理部门相关政策的制定,但并不具备任何法律效力。未来将基于新的研究结果对本共识进行不断地更新。
本共识制订专家委员会名单
学术顾问:
张学(哈尔滨医科大学)
执笔专家:
谢志颖(北京大学第一医院神经内科)
专家委员会成员(按姓氏汉语拼音排列):
陈万金(福建医科大学附属第一医院神经内科);
车薛华(复旦大学附属华山医院麻醉科);
崔丽英(北京协和医院神经科);
戴毅(北京协和医院神经科);
窦攀(北京大学第一医院临床营养科);
郭军红(山西医科大学第一医院神经内科);
洪道俊(南昌大学第一附属医院神经内科);
胡静(河北医科大学第三医院神经肌肉病科);
胡晓(北京大学第一医院麻醉科);
黄真(北京大学第一医院康复医学科);
李淳德(北京大学第一医院骨科);
李西华(复旦大学附属儿科医院神经科);
栾兴华(上海交通大学医学院附属第六人民医院神经内科);
马为(北京大学第一医院心血管内科);
马燕红(上海交通大学医学院附属第六人民医院康复医学科);
马祎楠(北京大学第一医院实验中心);
秦伦(北京大学第一医院康复医学科);
阙呈立(北京大学第一医院呼吸与危重症医学科);
商慧芳(四川大学华西医院神经内科);
史录文(北京大学医学部药学院);
宋雷(中国医学科学院阜外医院心肌病科);
宋学琴(河北医科大学第二医院神经内科);
王柠(福建医科大学附属第一医院神经内科);
王朝霞(北京大学第一医院神经内科);
吴丽文(湖南省儿童医院神经内科);
吴士文(解放军总医院第一医学中心神经内科);
谢志颖(北京大学第一医院神经内科);
熊晖(首都医科大学附属北京儿童医院神经内科);
焉传祝(山东大学齐鲁医院神经内科);
姚生(解放军总医院第六医学中心神经内科);
袁云(北京大学第一医院神经内科 罕见病医学中心);
于雪凡(吉林大学白求恩第一医院神经内科);
朱雯华(复旦大学附属华山医院神经内科);
张成(中山大学附属第一医院神经内科);
张巍(北京大学第一医院神经内科 罕见病医学中心);
张学(哈尔滨医科大学)


















