各省、自治区、直辖市及新疆生产建设兵团卫生健康委:
为进一步提高罕见病诊疗规范化水平,保障医疗质量安全,我委组织对《第二批罕见病目录》中软骨发育不全等86个病种分别制定了诊疗指南(见附件,可在国家卫生健康委网站医政司栏目下载)。现印发给你们,请各地卫生健康行政部门做好组织实施工作。
国家卫生健康委办公厅 2025年6月17日
软骨发育不全
软骨发育不全(achondroplasia,ACH)是一种导致非匀称性身材矮小的遗传性疾病。
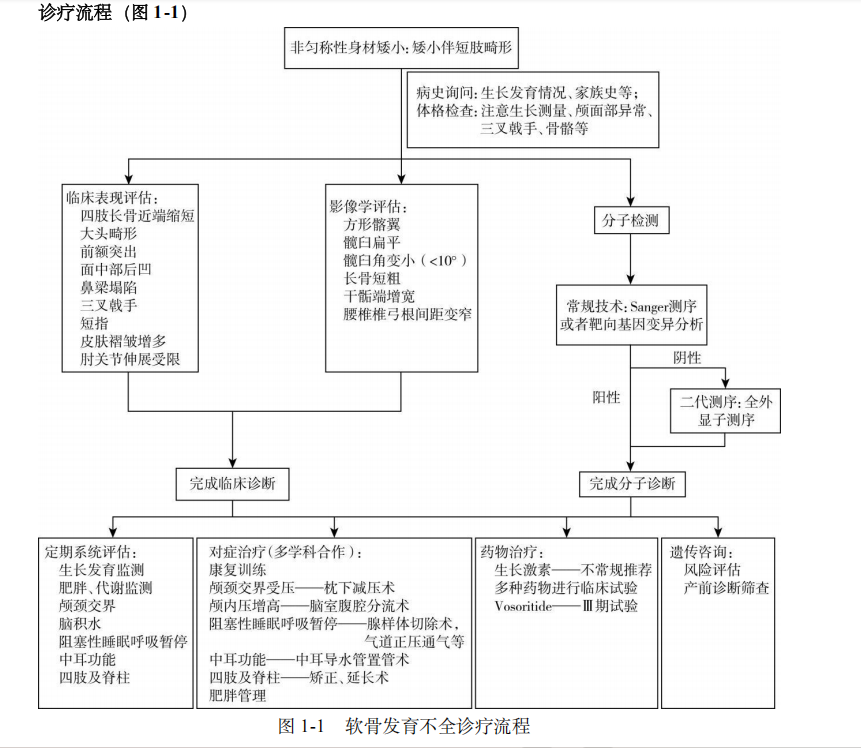
其典型的临床表现为四肢长骨近端短缩、大头畸形、三叉戟手以及特殊面容(前额突出、面中部发育不良呈现后凹和鼻梁塌陷)。
病因和流行病学
ACH遵循常染色体显性遗传规律,其主要致病基因是成纤维细胞生长因子受体 3(fibroblast growth factor receptor 3 ,FGFR3 )基因(OMIM#134934 )。其编码的FGFR3蛋白为人类四种成纤维细胞生长因子受体之一,普遍存在于软骨细胞表面,作为一种负性调节剂抑制软骨矿化,影响长骨的生长发育。当FGFR3基因发生功能获得性变异时,抑制软骨化骨,导致短肢畸形性身材矮小。大约 80%的病例源于新发变异,其发生与父亲年龄偏大存在一定关联,另外约 20%是由家族遗传所致。约98%的患儿为FGFR3基因c.1138G>A变异,少数为c.1138G>C变异,均导致p.Gly380Arg氨基酸改变,其外显率 100%。
国外报道,ACH在新生儿中的患病率为(3.72~4.60)/10万。我国ACH的患病率有待进一步的流行病学调查。
临床表现
ACH的典型临床表现包括:
①非匀称性身材矮小:患儿存在中至重度的身材矮小,并伴有短肢畸形。ACH男性成年后的平均身高约为130cm,而女性则约为124cm;
②颅面部发育异常:患儿表现为大头畸形,同时伴有前额突出、面中部发育不良呈现后凹、鼻梁塌陷;
③骨、关节及肌肉异常:患儿四肢长骨近端缩短,伴皮肤褶皱增多。此外,肘关节的伸展和旋转功能受限,伴有短指、三叉手、膝内翻等。
ACH的并发症相当广泛,主要有运动与语言发育迟缓、颅颈交界区狭窄常引起脊髓受压、脑积水、限制性肺疾病、阻塞性睡眠呼吸暂停、腰椎前凸、椎管狭窄、关节松弛、外侧半月板异常、中耳功能障碍、肥胖、心血管疾病、黑棘皮症等。
辅助检查
1.影像学检查 特征性表现包括短而坚固的管状骨;方形髂翼;扁平、水平移动的髋臼,髋臼角变小(一般小于 10°),坐骨切迹明显变窄;典型的股骨近端透亮度增加,干骺端增宽;尾椎椎弓根间距变窄;指骨近端和中端短;颅骨底部缩短;枕骨大孔、椎管狭窄,相应脊髓受压变性;侧脑室扩大等。
2.基因检测FGFR3基因第1138位核苷酸致病性变异,导致第380位氨基酸改变。
诊断
临床诊断主要基于疾病表现和影像学特点。诊断依据主要包括:不匀称性身材矮小、前额突出的大头畸形、中面部后缩、鼻梁凹陷、四肢近端缩短、皮肤褶皱增多、肘部伸展受限、短指、三叉戟手、膝内翻等特征性体貌,结合骨骼影像学表现,即可做出临床诊断。需进行骨骼X线、头颅脊柱MRI、多导睡眠监测、发育评估等来评估疾病的严重程度。
基因检测Sanger测序或靶向基因变异分析是常规采用的分子检测手段。对于那些用Sanger测序无法在常见变异位点发现变异,或者需要与ACH进行鉴别诊断的患儿,可以采用二代测序方法如全外显子测序进行检测。
鉴别诊断
一般而言,所有短肢性侏儒症都属于ACH的鉴别诊断范围。大部分短肢性侏儒症可以通过临床特征、影像学特征和表型出现的时间等进行直接区分。然而在少数情况下,这些疾病的临床表现可能较为相似,难以仅凭上述方法准确鉴别,需基因检测来进一步鉴别。
1.软骨发育低下症(Hypochondroplasia,HCH) 主要是由于FGFR3基因N540K或I538V变异所致,临床鉴别较困难,主要靠基因诊断区分。
2.致死性软骨发育不良症(Thana tophoric dysplasia,TD) 临床和影像学特征均与ACH类似,但程度要严重得多。均由FGFR3基因变异引起,但位点不同,故可依靠分子诊断区分。
3.SADDAN 综 合 征 ( Severe Achondroplasia with Developmenta l Delay andAcanthosis Nigricans,SADDAN) 严重ACH伴发育迟缓和黑棘皮病,是由于 FGFR3基因上第1949位碱基A>T的变异,引起蛋白K650M变异,需借助分子评估来准确地鉴别。
4.其他需要鉴别的疾病 软骨-毛发发育不良、假性ACH、其他干骺端发育不良疾病等。
治疗
已获批药物:已知FGFR3基因变异是ACH的致病原因,针对ACH分子病因的药物开发策略包括拦截FGFR3蛋白配体,阻断FGFR3蛋白,以及酪氨酸激酶抑制剂(FGFR3 蛋白受体的胞内成分),这些候选药物目前大多仍处于临床前研究及临床试验阶段。其中Vosoritide是一种 C 型钠尿肽 (CNP) 类似物,最近被批准用于软骨发育不全儿童从5岁到生长板闭合的促生长治疗。(vosoritide适应症已更新,美国全年龄段,欧盟>4个月,详情药物请查看下方相关阅读)
对于症状及并发症给予对症治疗。
重组人生长激素和肢体延长手术曾被尝试用于改善ACH患者的身高,但鉴于其疗效不确定以及潜在的副作用,这两种方法并不作为常规推荐。
对于颅内压增高患儿,可行脑室腹腔分流术;若颅颈交界区受压,可行枕下减压术。
出现严重阻塞性睡眠呼吸暂停时,治疗措施包括切除增生的腺样体或扁桃体、气道正压通气以及气管切开;针对中耳功能障碍,可行鼓膜切开置管术置入压力平衡。
此外,对于出现进行性下肢弯曲、脊柱后凸或椎管狭窄的患者,需行手术治疗。
临床管理
对于临床确诊为ACH的患儿,定期随访和系统评估至关重要,以及时了解其生长发育状况及并发症,必要时给予医疗干预。临床管理包括以下方面:
①生长发育监测:应用软骨发育不全标准化生长曲线监测;
②颅颈交界区监测:定期行头颈部神经影像学检查及神经系统检查;
③阻塞性睡眠呼吸暂停管理:注意关注睡眠中呼吸紊乱的症状和体征,必要时行多导睡眠监测;
④中耳功能监测:每年行听力测试,直至学龄前阶段;
⑤脊柱监测:对于3岁之前的患儿,建议每6个月进行一次脊柱评估。鉴于ACH成人患者患椎管狭窄风险较高,建议每3~5年进行一次神经系统检查,或在出现新的症状时及时评估;
⑥膝内翻监测:如果出现进行性疼痛或严重畸形,应转诊至骨科进行专业评估和治疗。
遗传咨询及产前筛查的建议
对于所有ACH患者及其家庭成员,应提供必要的遗传咨询服务,并对存在高风险的胎儿进行产前诊断。ACH的产前筛查包括绒毛或者羊膜腔穿刺,并同时结合超声检查来进行综合评估。在特定情况下,如患者有生育计划、患者和配偶同患ACH,或患者的配偶患有其他单基因遗传病等情况时,可以考虑植入前遗传学诊断技术来进行产前诊断。