导语
这一期【真知不罕见】,我们来聊一聊肌萎缩侧索硬化症(Amyotrophic Lateral Sclerosis, ALS)。
ALS的另外一个名称是大家所熟知的“渐冻人症”——这个病名的由来,是因为患者在确诊后,身体会逐渐丧失运动功能,彷佛被“冻住”了一样。
目前全球有超过5万5千名ALS病人。虽然ALS的病程发展各异,但多数患者在确诊后生存时间仅为3至5年,最终往往因呼吸肌无力导致的呼吸衰竭而去世。
实际上,早年曾非常火热的“冰桶挑战”公益活动就来源于这一疾病。而不幸罹患ASL的社会名人并不在少数,比如史蒂芬·霍金;蔡磊;张定宇等等……
渐冻人症是一种严重的神经退行性疾病,其主要病理特征是大脑和脊髓中的上、下运动神经元逐渐退化。随着神经元的退化,患者的四肢、躯干和肌肉出现进行性萎缩、无力,并最终完全丧失自主运动功能。
ALS的病理机制非常复杂,其发病涉及多个机制的相互作用。
首先,ALS的病因研究中,遗传因素是关键的一环,多个基因突变直接参与病理进程。这些基因突变可引发一系列细胞损伤反应,包括氧化应激的增加和过量的活性氧自由基在细胞中积累,这不仅会损伤蛋白质、核酸、脂质,还会破坏神经元的线粒体功能,进一步加剧细胞损伤。
同时,突变的基因还可能导致神经元的过度兴奋,使细胞内钙离子水平失衡,引发内质网和线粒体的应激反应。此外,蛋白质稳态失调,即异常蛋白的聚集与降解失调,往往阻碍细胞自噬和蛋白酶体降解系统的正常功能,形成毒性聚集体。
而在这一过程中,神经性炎症通过激活星形胶质细胞和小胶质细胞,释放炎症因子,加剧了神经元的退化和死亡。在没有已知基因突变的患者中,氧化应激、神经元过度兴奋、线粒体损伤、蛋白质稳态紊乱和神经性炎症这些机制依然在发挥作用。
综上所述,ALS的发病机制可以理解为多种病理因素共同参与的复杂网络,遗传因素和非遗传因素相互影响并共同推动疾病发展(图一)。
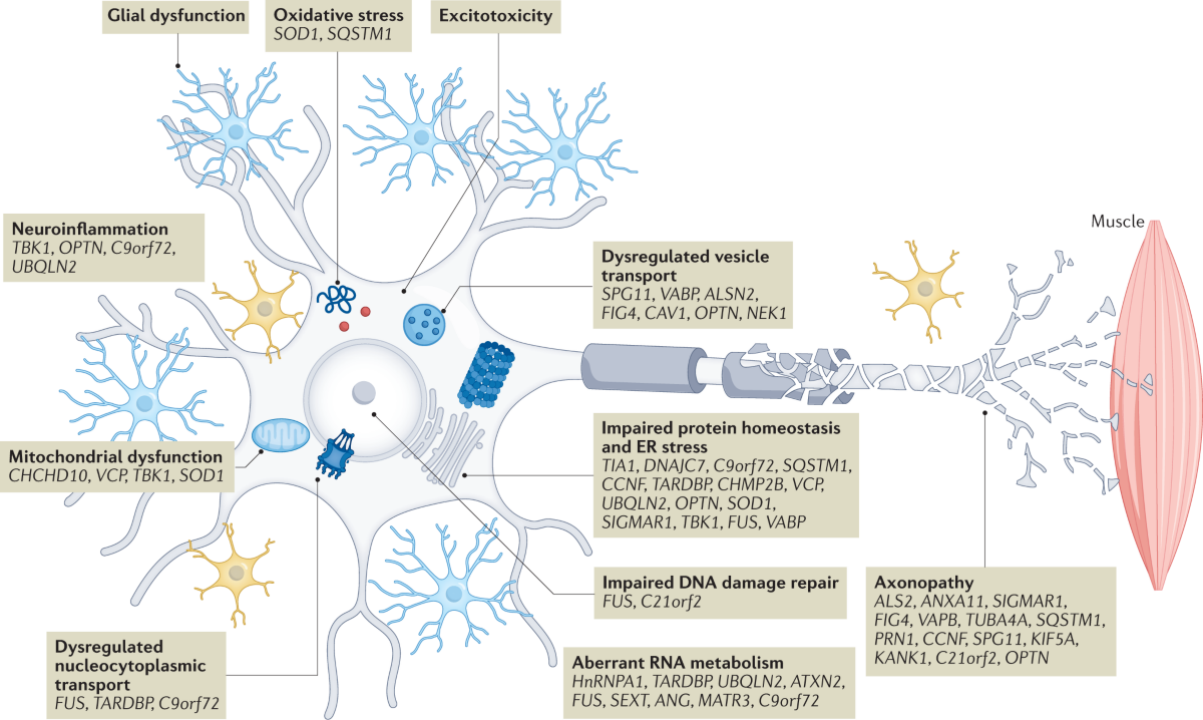
图一. ALS的病理生理学、遗传因素和风险(来自Pamela J. Shaw等作者的Nature Reviews Drug Discovery文章)
考虑到该疾病的致病因素比较复杂,治疗药物和治疗手段也备受关注,本期以渐冻人症为主题的【真知不罕见】将分为“上、中、下”三篇。
本篇为上,着重讲述遗传因素对渐冻人症的影响。
遗传因素:基因如何影响ALS风险?
自1993年,Robert Brown团队(现位于美国麻省大学陈医学院)发现了首个与ALS相关的致病基因SOD1以来,ALS的遗传学研究取得了飞跃性进展。ALS的遗传背景显示了高度异质性,即便是散发性ALS (sporadic ALS, sALS) 患者也可能携带致病基因突变。ALS可分为家族性ALS(familial ALS,fALS,占5-10%)和散发性ALS (sALS,占90-95%)。虽然散发性ALS患者通常没有家族史,但大基因组相关性分析(genome-wide associatioin studies, GWAS)发现,多数sALS患者也携带与ALS发病相关的罕见基因突变。
在已经被诊断出的30多个致病或高风险基因中(图一),四个最常见的ALS致病基因是C9ORF72、SOD1、TARDBP和FUS,这些基因突变占据了大约70%的欧洲家族性ALS患者病例。
SOD1
SOD1基因是第一个被发现的与ALS相关的致病基因,它编码超氧化物歧化酶1(Superoxide dismutase 1, SOD1),这是一种抗氧化酶,负责清除细胞中的氧自由基。SOD1基因突变是家族性ALS的主要致病原因之一,约占家族性ALS的20%,在散发性ALS中也占1-2%。迄今为止,已发现超过18种SOD1基因突变。这些突变大多是点突变,导致SOD1蛋白的结构和功能发生异常,例如G93A突变是SOD1的第93个氨基酸甘氨酸(Gly, G)替换为丙氨酸(Ala, A),破坏了SOD1蛋白的正常折叠和稳定性。当SOD1基因发生突变时,产生的异常蛋白会形成不溶性聚集体,聚积在神经元中,导致细胞应激反应和神经元死亡。SOD1突变导致的ALS通常在成年期发病,值得一提的是,患者通常不伴随认知功能障碍。不同SOD1突变的临床表现也存在一定的异质性,某些突变可能导致较为缓慢的病程,而一些突变如A4V(SOD1的第四个氨基酸丙氨酸替换为缬氨酸)则表现出极为迅速的进展,患者生存期可能仅为1-2年。
C9ORF72
C9ORF72突变是目前已知最常见的ALS致病基因,尤其在家族性ALS患者中,约40%的患者携带这一突变。C9ORF72基因的异常表现为重复序列GGGGCC的扩增,正常人群C9ORF72基因的一号内含子中GGGGCC重复序列通常少于30个,而ALS患者的重复序列可多达数百至数千次。这一异常扩增会导致多方面的分子紊乱,显著影响神经元功能。
首先,这些异常的重复序列产生的正义链(GGGGCC)和反义链(GGCCCC)RNA会影响C9ORF72蛋白质的表达。重复的RNA结构降低了RNA的稳定性,并可能减少蛋白质的翻译效率,从而导致C9ORF72蛋白的表达水平下降,破坏其在细胞内的正常功能。其次,这些扩增的重复RNA序列会募集关键的RNA结合蛋白,导致这些蛋白的正常浓度下降,进而干扰细胞内的RNA代谢和其他相关过程。这些蛋白功能的丧失可能破坏神经元的正常活动,进一步加剧疾病发展。此外,正义和反义RNA通过一种称为重复序列相关的非ATG起始翻译(RAN)机制生成有毒的双肽重复蛋白(dipeptide repeat protein, DPR),包括正义链生成的GR,GA和GP多肽以及反义链生成的GP,PR和PA多肽。这些双肽重复蛋白在C9ORF72-ALS患者的病变组织中被广泛发现,并并证明具有神经毒性,直接损害神经元的健康和存活。
C9ORF72基因位于9号染色体的72号开放阅读框,突变通常表现为显性遗传,多数患者在成年期发病。C9ORF72蛋白质在RNA处理和代谢中发挥重要作用,而重复序列扩增所引起的蛋白质表达和功能紊乱,对ALS患者的神经系统具有深远的影响。C9ORF72基因突变的患者还可能伴随额颞叶失智症(frontotemporal dementia, FTD),进一步加重神经系统的损伤。
TARDBP
TARDBP基因编码的TDP-43 (TAR DNA-binding protein 43) 蛋白在正常情况下主要位于细胞核中,负责调控RNA代谢过程,包括转录,剪切,mRNA运输和稳定性。然而,在ALS患者中,TDP-43蛋白出现异常,发生核-质易位并在细胞质中形成不溶性聚集体。这种聚积和易位被认为是ALS病理学的核心特征之一。虽然TARDBP基因突变仅在少数ALS患者中存在,但97%以上的ALS患者表现出TDP-43的异常积累,因此TDP-43异常在ALS中具有广泛的影响力。
TDP-43的异常易位和聚积对神经元造成多方面的损伤。首先,TDP-43离开细胞核后,其在RNA代谢中的关键功能受损,导致特定基因(如UNC13A)的异常剪接和表达,破坏神经元的转录组平衡。此外,细胞质中聚积的TDP-43形成毒性蛋白包涵体,干扰细胞自噬和蛋白质降解,甚至影响线粒体功能并加剧氧化应激,损害神经元的生存。
FUS
FUS基因编码的一种RNA结合蛋白(Fused in sarcoma RNA binding protein)在RNA代谢中发挥关键作用,包括RNA的剪接、运输和转录调控。该蛋白的突变在ALS病理中起着重要作用,与TDP-43类似,正常生理状态下,FUS蛋白位于细胞核内,但由于FUS基因突变,FUS蛋白从核易位到质,并在细胞质中形成不溶性聚集体。这种聚积体不仅破坏了FUS蛋白的正常功能,还会干扰神经元的其他细胞过程,导致神经元的损伤和退化。
虽然FUS基因突变较为罕见,但其导致的ALS病情通常较为严重,尤其是早发型的病例。患者往往病程短,病情进展速度快,生存期较其他ALS类型更短。但与其他ALS类型不同的是,这类患者的认知功能一般不受影响。
ALS的遗传因素研究揭示了多种致病基因,这些基因突变涉及RNA代谢、蛋白质聚集和神经元健康等多个方面,导致神经元功能异常和退化。此外,部分ALS基因还与其他神经退行性疾病(如额颞叶失智症)存在重叠,表明它们可能共享病理机制。在下期内容中,我们将详细介绍除了基因突变带来的影响外,氧化应激、神经元过度兴奋、线粒体损伤、蛋白质稳态失调和神经炎症等非遗传因素在ALS的病程中的重要作用。
注释:
大规模基因组分析揭示了多种导致ALS的致病基因和风险因素,这些基因变异不仅参与了多种运动神经元退化的关键病理机制,还与相邻细胞如胶质细胞和中间神经元有关。
Glial dysfunction: 胶质细胞功能障碍
Oxidative stress: 氧化应激
Excitotocixity: 神经元兴奋毒性
Neuroinflammation: 神经炎症
Mitochondrial dysfunction: 线粒体功能失调
Dysregulated nucleocytoplasmic transport: 核-质转运失调
Dysregulated vesicle transport: 囊泡运输失调
Impaired protein homeostasis and ER stress: 蛋白质稳态失调和内质网应激
Impaired DNA damage repair: DNA损伤修复受损
Aberrant RNA metabolism: RNA代谢失常
Axonopathy: 轴突病变
撰文 | 杨惠雅
审核 | 喻柏雅


















