罗氏(Roche)近日在法国埃夫里举行的第二届国际脊髓性肌萎缩症科学和临床大会上公布了脊髓性肌萎缩症(SMA)口服药物risdiplam III期SUNFISH试验关键第二部分(Part 2)的一年期数据。SUNFISH是有史以来对2型或3型SMA患者(2-25岁)进行的最大规模的安慰剂对照试验,第二部分研究群体代表了广泛的、现实世界SMA人群,结果显示:治疗一年后,与安慰剂组患者相比,risdiplam治疗的患者运动功能表现出显著改善。值得一提的是,SUNFISH是纳入SMA成人患者的首个安慰剂对照试验,证实risdiplam改善或稳定了运动功能。
risdiplam是一种运动神经元生存基因2(SMN2)剪接修饰剂,开发用于所有类型(1型、2型、3型)SMA的治疗。目前,该药正在接受美国FDA的优先审查,该机构将在今年5月做出审查决定。如果获得批准,risdiplam将成为治疗所有3种类型SMA的首个口服药物。
SUNFISH是一项两部分、双盲、安慰剂对照的关键性临床研究,在2型或3型SMA儿童和年轻成人(2-25岁)患者中开展。第一部分确定了验证性第二部分的剂量,探索性终点是评估疗效。第二部分是一项大型安慰剂对照试验,评估risdiplam治疗2型或3型SMA患者。
结果显示,治疗一年后,与安慰剂组相比,risdiplam治疗组患者运动功能表现出显著改善,在主要终点和关键次要终点方面取得了具有医学意义和统计学意义的结果。主要终点方面,与安慰剂组相比,risdiplam治疗组患者运动功能测定量表(MFM-32)评分相对基线的变化显著更大(平均差异:1.55分,p=0.0156)。关键次要终点方面,与安慰剂组相比,risdiplam治疗组患者在修订上肢模块(RULM)测量的上肢功能方面也显示出改善(差异:1.59分,p=0.0028)。
正如预期的那样,探索性亚组分析显示,在最小年龄组(2-5岁)中观察到了risdiplam相对于安慰剂的MFM-32最强应答(78.1% vs 52.9%达到≥3分增加)。重要的是,在18-25岁年龄组中观察到疾病稳定(57.1% vs 37.5%,稳定定义为≥0分增加),这是存在更多疾病的患者的一个治疗目标。
该研究中risdiplam的安全性与已知的安全性一致,没有发现新的安全信号。risdiplam不良事件概况与安慰剂相似。迄今为止,已有400多例患者在多项试验中接受了risdiplam治疗,在任何一项试验中均没有导致研究停药的治疗相关安全性发现。
SUNFISH首席研究员、意大利天主教大学儿科神经病学系医学博士Eugenio Mercuri表示:“risdiplam是在包括儿童、青少年和成人在内的广泛SMA患者群体中获得关键安慰剂对照数据的第一个潜在疗法。数据表明,通过改善2型或卧床3型SMA患者的运动功能,risdiplam可保持和潜在地提高患者的独立性。”
罗氏首席医疗官、全球产品开发主管、医学博士Levi Garraway表示:“我们对在这一广泛的SMA患者群体中取得的积极结果感到非常鼓舞,这些患者中的许多在临床试验中代表性不足。这些研究有助于我们了解哪些测量指标与患者最为相关,以及稳定病情对已确诊疾病患者的重要性。”
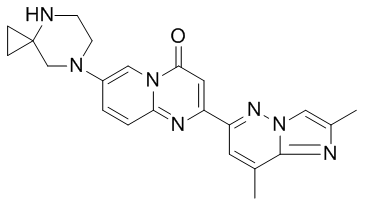
risdiplam化学结构(图片来源:medchemexpress.cn)
risdiplam是一种口服液体、运动神经元存活基因2(SMN2)剪接修饰剂,旨在持续增加和维持中枢神经系统和外周组织中的SMN蛋白水平。越来越多的临床证明表明,SMA是一种多系统疾病,SMA蛋白的丢失可能影响中枢神经系统以外的许多组织和细胞。risdiplam口服给药后呈现全身性分布,可持续增加中枢神经系统和外周组织的SMN蛋白水平,已显示出可改善1型、2型、3型SMA患者的运动功能。
作为与SMA基金会及PTC Therapeutics公司合作的一部分,罗氏领导了risdiplam的临床开发项目,该公司正在开展4项全球性多中心临床研究(SUNFISH[NCT02908685]、FIREFISH[NCT02913482]、JEWELFISH[NCT03032172]、RAINBOWFISH[NCT03779334]),评估risdiplam治疗所有类型(1型、2型、3型)SMA以及新生儿症状前SMA的疗效和安全性。
2019年11月,美国食品和药物管理局(FDA)受理了risdiplam的新药申请(NDA)并授予了优先审查,并已指定处方药用户收费法(PDUFA)目标日期为2020年5月24日。之前,FDA已授予risdiplam孤儿药资格和快速通道资格。如果获得批准,risdiplam将成为治疗所有3种类型SMA的首个口服药物。
SMA治疗:全球已有2款药物上市,Spinraza于2019年2月在中国获批
SMA是一种会导致肌肉无力和萎缩的运动神经元性疾病,该病属于基因缺陷导致的常染色体隐性遗传病,对患者周身上下的肌肉都会造成侵害,患者主要表现为全身肌肉萎缩无力,身体逐渐丧失各种运动功能,甚至是呼吸和吞咽。SMA是2岁以下婴幼儿群体中的头号遗传病杀手,该病是一种相对常见的“罕见病”,在新生儿中的患病率为1:6000-1:10000。据相关报道,目前中国SMA患者人数大约3-5万人。
2016年12月,来自渤健与合作伙伴Ionis开发的药物Spinraza(nusinersen)获批,成为全球首个治疗SMA的药物。该药是一种反义寡核苷酸(ASO),通过鞘内注射给药,将药物直接递送至脊髓周围的脑脊液(CSF)中,改变SMN2前信使RNA(pre-mRNA)的剪接,增加全功能性SMN蛋白的产生。在SMA患者中,SMN蛋白水平不足导致脊髓运动神经元功能退化。在临床研究中,Spinraza治疗显著提高了SMA患者的运动机能。
2019年5月,来自诺华的基因疗法Zolgensma(onasemnogene abeparvovec)获批,成为全球首个治疗SMA的基因疗法。该药通过单次、一次性静脉输注后持续表达SMN蛋白来阻止疾病进程,可解决SMA的根本病因,有望长期改善患者生存质量。
在中国市场,Spinraza于2019年2月底获批,用于5q脊髓性肌萎缩症(5q-SMA)患者的治疗。此次批准,使Spinraza成为中国市场首个治疗SMA的药物。5q-SMA是SMA的最常见类型,约占全部SMA病例的95%,该类型SMA是由5号染色体上的SMN1(运动神经元生存蛋白1)基因突变所引起的,因此得名5q-SMA。
原文出处:Roche’s risdiplam showed significant improvement in motor function in people aged 2-25 with type 2 or 3 spinal muscular atrophy


















