
小雅的绘画作品

空药盒堆成了小山
罕见病,因发病率低,治疗药物被称为“孤儿药”。由于患者少,在正常的市场条件下很难靠销售回收高额的研发投入,因此治疗罕见病的药难买且价高。
去年9月,腾讯发起的99公益日期间,一条《8岁小女孩买伟哥?妈妈含泪说出真相》的短视频火了。视频主人公小雅,罹患一种名叫肺动脉高压的罕见病,像她一样要靠“伟哥”类药物续命的这一群体,开始走入大众的视线范围,大河报曾对此事进行过关注报道。
3月24日,记者来到许昌小雅的家回访,小雅妈妈高兴地告诉记者:“本月17号,河南省医保局发文,将国家谈判药品的27种药品暂定为我省门诊特定药品,其中就有女儿的适用药!该政策4月1日起执行,以后就能少花些钱买到续命药啦!”
事例1 多方关爱“蓝嘴唇”孩子得到救助
噩梦降临,家里多了个“蓝嘴唇”孩子
3月24日,记者来到了河南省许昌市魏都区某庄小雅的家回访,小雅正在家中写作业、画画,精神十足。“孩子现在身体比之前好太多了!”小雅母亲尚女士告诉记者,3岁前的小雅,和别的孩子没什么不同。孩子3岁时突然没缘由地反复发烧、咳嗽,辗转了好多医院,做了很多检查,最终确诊为特发性肺动脉高压。
这是啥病?经过与医生沟通和自己查资料,尚女士才知道这种病被喻为“心血管癌症”,患病率低,为万分之一,目前尚无法治愈,仅能靠基础治疗,大部分患者活不过3年。患者外表健全,却行动受限。因长期缺氧,患者嘴唇常呈蓝紫色,所以“蓝嘴唇”是人们对肺动脉高压患者更通俗的称呼。
高昂的续命药费,几乎压垮这个家庭
“我愿把自己的肺换给女儿,哪怕是以命换命,但医生说这方案行不通。”尚女士说,小雅确诊后半年,病情进一步恶化,甚至连十几米的路都不能走,“她会喘着气捂着心口,说‘妈妈,我这里疼’,那是我这辈子生命里最黑暗的一段经历,我觉得我要失去她了……”
小雅患上的是肺动脉高压中最为棘手的一类:特发性肺动脉高压。找不到发病的确切原因,只能用药把肺动脉压“强压”下来。于是,尚女士和家人开始打起精神“拼命赚钱”。“小雅需要联合用药,不但要预防血管收缩,还要扩张血管。新研发上市的药特别贵,比如马普腾坦,一个月花费就要近3万元。”尚女士说,几年来家里辗转为孩子治疗已花费50余万,欠有7万多元外债,已无法负担孩子联合用药,最后甚至连一种药也无法做到足量服用。
尚女士从内间拎出两袋装满空药盒的塑料袋,“这些都是孩子吃的,小袋的是去年9月至今吃的,大袋的那些不记得是从何时开始吃的了。”两个塑料袋放在地上,药盒瞬间堆成“小山”……
一瓶伊米苷酶的价格是21850元
治疗肺动脉高压的波生坦等4种药品纳入医保目录
社会关注后,小雅得到了多方援助
因为肺动脉高压患者不能进行高强度运动,更不能劳累,所以小雅出门,大多数时候都是坐在妈妈拉的一辆简陋折叠小拖车上。但在此次采访中,记者发现小雅能跑跳嬉戏,甚至还能和相熟的腾讯公益平台的工作人员一起玩病友送她的滑板车。“去年9月我们来看小雅,她的嘴唇还是蓝紫色的,不太能活动,现在看她身体明显有了好转。”一位腾讯公益平台的工作人员告诉记者。
尚女士表示,由于媒体和腾讯公益平台的关注,去年9月,广州白云山医药销售有限公司一行工作人员来到家中,当天就带来了金戈(枸橼酸西地那非,俗称“伟哥”)280粒,并承诺提供3年的药品。华中阜外医院结构性心脏病病区主任医师程江涛教授协调江苏一家企业,免费提供一年的药物安立生坦。小雅又可以联合用药了。此外,广药白云山还协助小雅入住广州医科大学附属第一医院。去年12月5日,中国工程院院士、国家呼吸疾病临床医学研究中心主任钟南山教授带领院士团队为小雅进行了会诊。“本来这个3月我们应该去复查的,因疫情耽误了。我有信心,因为天天在照顾孩子,她的变化我都看在眼里,孩子的病情是在逐渐好转的!”尚女士说。
适用药进入河南省门诊特定药品行列
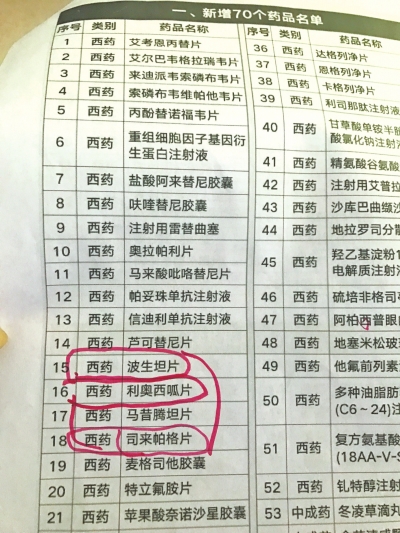
令尚女士更为高兴的是,近日,河南省医保局下发了《关于规范完善我省特重大疾病医疗保障门诊特定药品使用管理工作的通知》(下文简称《通知》),“通知将2019年国家谈判药品的27种药品暂定为我省门诊特定药品,其中就有我家孩子适用的药,这样一来我们就能以更便宜的价格买到续命药了!”尚女士称,她将此消息发到微信病友群中,有的病友家属甚至在群里放起虚拟烟花,尚女士边说边抹泪,“我这是高兴的!”
原来,去年11月底,国家医保局、人力资源社会保障部印发《关于将2019年谈判药品纳入〈国家基本医疗保险、工伤保险和生育保险药品目录〉乙类范围的通知》,治疗肺动脉高压的波生坦等4种药品纳入医保目录。“能进医保目录,药价已在谈判时被砍掉不少,等于算是‘打折’了!按河南医保局新下发的《通知》,小雅甚至不用住院,在门诊就能享受到部分药费医保报销!”尚女士说。
“希望媒体能多多关注罕见病患者这一群体,这个群体实在太难了!诊治难、买药难、进医保难,有的罕见病患者甚至等不到适用药进医保目录,人就不在了……”尚女士说。
事例2 罹患戈谢病的小姑娘,每月需要10万的药费来续命
一粒不幸的微尘,落在某个家庭里,就是绝对的不幸。
“1公斤,3277元,这不是空穴来风,这是我现在女儿生命的价格!”25日,戈谢病小患者开心(化名)的爸爸蒋先生哽咽着向记者讲述了女儿的遭遇。2012年12月,开心出生,“这个生命和我有关,我发誓要把这世间最好的都给她。可命运让我成了食言的人,就在开心7岁时,她确诊得了一种罕见病。”
蒋先生告诉记者,得了这种病的患者,肚子往往比正常人的要大,“就像我女儿这样,因为体内缺少一种酶,所以会导致身体内该被代谢掉的东西没办法代谢掉,从而堆积在患者的肝脏、脾脏等各个器官,如果不进行有效的治疗,只有死路一条”。
据了解,戈谢病的患病率是五十万分之一。“河南目前我知道的有28名患者,医生说,这种病虽罕见,但有一种叫伊米苷酶的药可用。只要按时按量用药,孩子就能像正常人一样活着。”蒋先生说,这种药的用药剂量,跟体重有关,每公斤要使用60个单位的药,一瓶药是400个单位的量,价格是21850元,每个月要用两次药。自己女儿每月药费10.488万元,全自费,需终身服用。
“听完这个结果,我一下蒙了,我攥着口袋里的银行卡,问自己,你的钱可以买女儿多久的命呢?我和妻子躲在墙角,抱头痛哭,从那一天,那一刻,我们家的天就塌了……作为一个父亲,我觉得自己特别无能,因为我没钱,因为我救不了女儿的命!”蒋先生哭道,所以,他不敢像其他父母一样,期盼孩子长高一点,长胖一点,“我们唯一希望的是,她能长慢一点,因为慢一点,她就能活得久一点。”
蒋先生说,他和妻子只是希望孩子能活下去,不管用什么方式,不管付出什么代价,“孩子想活着,孩子本可以活着。有什么办法能救救我的孩子?”
声音 “不罕见”的罕见病救助保障需多管齐下
“在我们科室的临床上,现在几乎每周都会遇到一例我们从未接触过的罕见病。”3月26日,河南中医药大学第一附属医院儿科遗传代谢内分泌专业学术带头人郑宏,在受访时表示,全球已知7000多种罕见病,中国罕见病患者大约在1680万,有80%是由于基因缺陷所导致的遗传病。
据郑宏介绍,脊髓性肌萎缩症在欧美人群存活新生儿中的发病率约为1/10000,携带者频率为1/40-50,在2岁以下儿童中居致死性遗传病的首位。目前中国尚无发病率的确切数据,中国人群中的携带者频率约为1/42。目前基因靶向治疗,这支“神药”单价为69.9万,“第一年需注射6次,以后每年4次。目前中国初级保健基金会在援助,负荷剂量的4支是买1支捐助3支,维持剂量是买1支捐助1支。第一年治疗药物费用是140万左右,以后每一年100万左右。这个费用对普通家庭来说,是天价。”
由于罕见病的特殊性,单个罕见病病种,无论在新药研发还是临床治疗都面临着诸多挑战。有不少罕见病认识水平低、误诊率高、药物可及性差。罕见病药物也面临着投入高、研发难、上市难的风险。所以,对于一些罕见病患者来说,无药可医、有药但价高难求、适用药难进医保,都是常态。
郑宏告诉记者,在河南,很多罕见病患者都在“抱团取暖”,“我们会给他们建微信群,有时是他们自发建群,大家可以沟通病情,也方便医生随诊。我手头现在管理有8个这样的群”。
在罕见病救助和保障方面,郑宏表示,我国各地方政府也正在摸索各种模式。一方面可将社会保险与慈善相结合,提高罕见病药报销比例;另外,也可针对罕见病医保报销制定出独立政策,单独筹资单独支付。“慈善机构可以整合社会各界的爱心力量。也可适当鼓励地方政府根据当地财力和慈善捐款的缺口情况,为罕见病慈善基金的发放提供配套资金支持。”


















