
背景介绍
过去十年间,基因组范围测序(GWS)——包括全外显子组测序和全基因组测序——已成为全球许多地区罕见遗传病(RGD)诊断的标准工具。根据适应症和具体实施方案的不同,估计约40%接受GWS检测的RGD家庭会获得明确诊断。相比之下,作为一线检测的染色体微阵列(CMA)在发育迟缓、畸形特征、智力障碍、学习障碍、自闭症及多发先天性异常等适应症中的平均诊断率仅为12.2%。然而,尽管临床GWS检测已取得显著成效,仍有相当比例的RGD家庭在临床检测后未能获得明确诊断。
在临床基因组范围测序(GWS)检测后未获得诊断的家庭,大致可分为三类:(1)其疾病的病因并非单基因所致,且不太可能通过当前遗传检测方法明确;(2)其疾病的遗传病因未被所采用的GWS方法检出(例如通过外显子测序时可能漏检复杂结构变异),但可能通过其他技术手段明确;(3)其疾病的遗传病因虽可通过GWS检出,但相关基因、区域或变异尚未被证实与疾病相关(例如由于分析挑战或证据不足)。换言之,最后一类家庭的现有测序数据中其实包含诊断信息,但在临床检测时尚无法出具报告。
鉴于每年新发现的疾病-基因关联和疾病-变异关联速度迅猛,学术界对重新分析临床产生的基因组范围测序(GWS)数据以寻找RGDs未检出的分子病因抱有极大兴趣。许多大型RGD研究项目——如未确诊疾病网络(美国,https://undiagnosed.hms.harvard.edu/)、Genomic Answers for Kids(美国,https://www.childrensmercy.org/childrens-mercy-research-institute/studies-and-trials/genomic-answers-for-kids/)、RD-Connect(欧盟,https://rd-connect.eu/)以及本文讨论的Care4Rare(加拿大,https://www.care4rare.ca/)——均采用这一策略,即对临床产生的GWS数据进行“再分析”,以寻找RGDs未被识别的分子病因。对涉及临床GWS数据再分析的文献综述显示,新的诊断率中位数为15%,但各研究间差异显著。在科研背景下处理GWS数据,可拓宽用于识别RGD分子病因的工具和方法。我们近期对一组287例临床GWS检测后未确诊的家庭进行了“临床再分析”。该再分析仅限于在线孟德尔遗传数据库(OMIM)中已知与疾病相关的基因变异,结果在39个家庭(14%)中发现了具有说服力的候选变异,并最终为13个家庭(5%)确诊。促成这些新诊断的最常见因素是新的基因组知识的可用性,包括新的疾病-基因关联、疾病-变异关联以及现有疾病表型的扩展。这些发现表明,如果这些数据随时间推移被重新分析,部分未确诊的RGD家庭将因全球基因组学知识的进步而获得诊断。
尽管定期临床再分析可能捕获新发现的疾病关联中的诊断案例,但无法实时促进数千种新型疾病-基因关联的发现。对于病因不明的RGD家庭,其病情将始终无法确诊,直至有足够证据将其特定疾病与致病基因相关联。证实此类关联的重要标准是:在相同基因中发现多个无亲缘关系的先证者携带致病性变异,且表型重叠,以致被认为患有同一种未描述的疾病。这种识别相似家庭的过程通常被称为基因组配对。由于RGDs本质上极为罕见,新疾病基因的发现依赖于全球范围内未确诊家庭信息的共享。为此,Matchmaker Exchange(MME)等工具使全球提交者(可能是研究人员、临床医生或RGD家庭)能够连接并讨论病例潜在的重叠。MME采用双向配对策略,只有当提交者各自家庭标记了相同的新候选基因(即假设存在新型疾病-基因关联的基因)时才会进行匹配。自2015年成立以来,MME已被研究和临床罕见病社区广泛用于促进数百种新型疾病-基因关联的发现。Care4Rare通过MME进行双向配对的实践经验印证了其效用:仅2年内,Care4Rare就匹配了194个新候选基因,与其他提交者建立了861次连接,最终促成其中23个基因(15%)的合作研究。
尽管双向配对一直是基因发现的基础工具,但该方法的可及性受到多重限制。首先,这种基于假设的匹配要求每个家庭在提交前必须标记新候选基因(即必须经过分析/审查)。其次,虽然当前连接至MME的大多数数据库支持在提交新候选基因时纳入参与者级信息(如特定变异、合子状态、详细表型和遗传模式),但根据我们的经验,大多数初始匹配案例缺乏这些信息。因此,关于潜在匹配的额外细节必须在初始匹配后通过邮件交换,这对用户而言耗时费力。现有的双向配对模型因而将可共享数据限制在满足以下条件的少数家庭:(i) 已进行(重新)分析且(ii) 标记有新候选基因,同时将提交者限制为具备资源通过邮件跟进大量潜在匹配的群体。鉴于临床和科研检测产生的海量GWS数据,必然存在大量未开发的RGD数据集尚未纳入双向配对,这阻碍了为未确诊家庭识别新型遗传病因的能力。
为弥补双向配对的局限性,学界已提出多种基因组配对新方法。单向配对模式由单方发起查询,通过向基因组范围测序(GWS)数据库提交目标基因或变异信息,筛选携带匹配变异的未确诊参与者。尽管尚未建立类似MME的RGD数据库网络用于单向配对,但多个数据库已自主开发实现方案。MyGene2、Geno2MP、VariantMatcher和Franklin采用变异级单向配对,允许用户检索特定变异在数据库中的存在情况,并获取携带该变异参与者的表型信息。而DECIPHER、RD-Connect GPAP和seqr等平台则支持基因级单向配对,用户可查询目标基因并获得该基因所有变异。评估现有单向配对方案后,我们认为基因级方法具有通过查询两类基因识别未确诊RGD家庭目标变异的潜力:(1)新近发现与人类疾病相关的基因;(2)尚未建立疾病关联的基因。鉴于基因级方法可能根据数据库规模返回大量变异,我们预期需要补充相关数据以排除假阳性匹配。然而据我们所知,目前尚未评估单向配对用户过滤基因级查询所需的数据类型和详细程度,以将潜在目标变异数量控制在可管理范围。
本文介绍了一种基因级单向配对的工作流程,以及支持该方法的工具测试版——单向配对门户(OSMP)。该平台设计重点在于提供可定制界面中的多种参与者级和变异级信息,以最佳支持用户进行高效的单向配对查询,并限制筛选潜在匹配所需的外部沟通。为测试OSMP及其单向配对方法的实用性,我们使用两组基因对Care4Rare注册参与者中的新目标变异进行了测试研究:(1)130个新描述的OMIM疾病基因,用于搜索在上次GWS分析时未被识别的致病性变异;(2)178个由Care4Rare项目在GWS分析中标记的新候选基因。
材料与方法
我们设计并开发了一款名为OSMP的基于网络的门户工具,该工具可连接一个或多个包含研究参与者变异与健康信息的RGD数据库。OSMP测试版支持基于基因的PhenoTips实例查询,通过从PhenoTips变异存储库获取并显示单核苷酸变异(SNVs)和小插入/缺失(indels),同时从PhenoTips参与者记录中提取参与者信息[20]。OSMP前端采用React JavaScript库开发,后端基于Node.js框架设计。用户认证通过Keycloak服务器管理,支持使用连接RGD数据库的凭证实现单点登录。
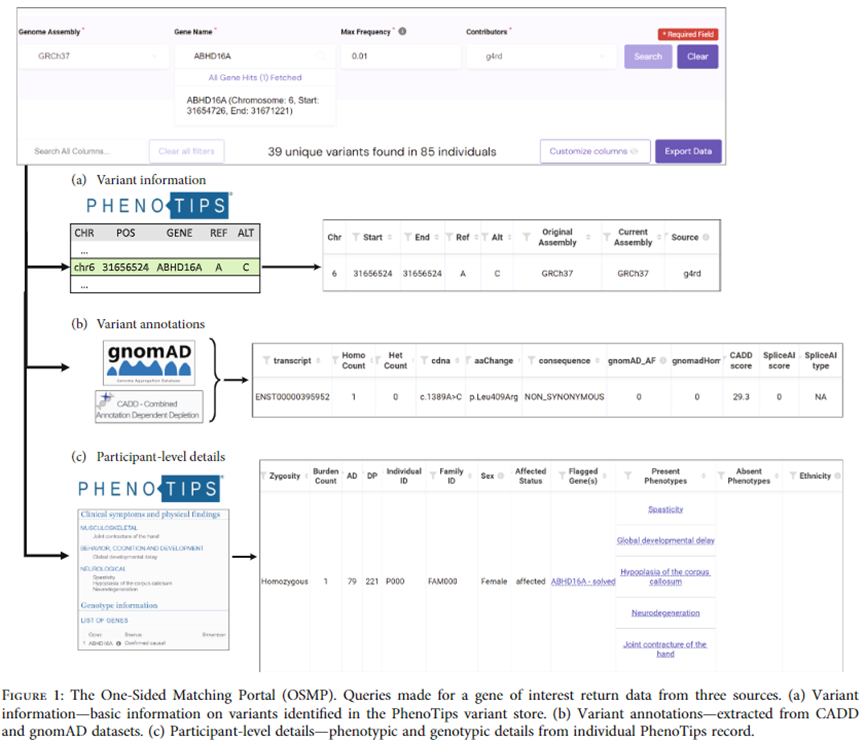
基于基因的单向匹配预计会产生大量潜在匹配结果,因为它会返回数据库中所有参与者(患病与未患病)针对特定基因存在的所有变异观察(VOs)。因此我们设计了一套工作流程,通过七步筛选将大量返回的VOs缩减为更小、更可能致病的候选列表(图2)。该工作流程旨在优先筛选出最可能影响蛋白质功能、且符合查询基因在表型重叠参与者中遗传模式的VOs。
结果
我们首次对单向匹配工作流程进行测试测试,通过查询一组近期与疾病关联的OMIM基因(即OMIM基因测试)来验证OSMP的功能。我们假设,由于这些疾病-基因关联在参与者最近一次全基因组测序分析时尚未被报道,该基因集可能富含未解决的RGD患者中的致病性变异。首先,我们生成了2021年11月至2022年8月期间新增至OMIM的疾病-基因关联列表(n=227)。通过人工审查,优先筛选出临床表现与Genomics4RD患者群体最相关的疾病(即严重影响单个或多个系统的疾病),最终得到145个基因的列表。随后,根据OSMP当前20万碱基对的内存限制,进一步筛选出基因大小符合条件的关联(剔除15个基因),最终获得130个疾病-基因关联(涉及116个独特基因,其中14个基因具有两个新描述的疾病关联)。使用OSMP查询这些关联时,约63%(82/130)为常染色体隐性,32%(42/130)为常染色体显性,5%(6/130)为X连锁隐性。
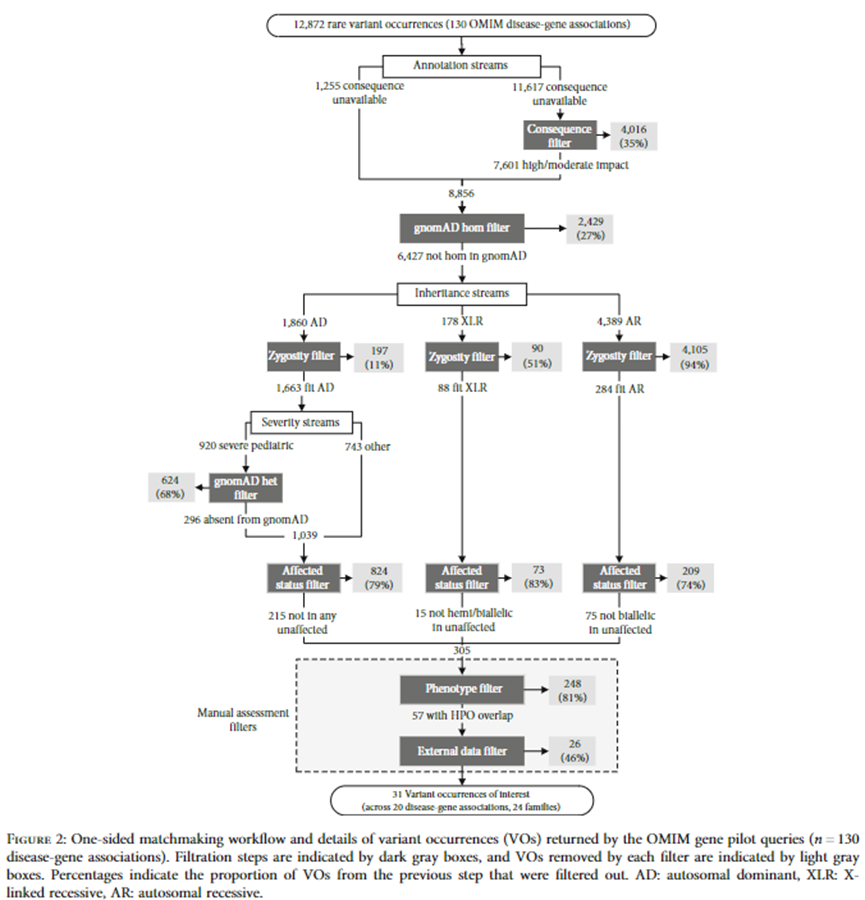
OSMP在查询的130个疾病-基因关联中,共返回了12,872个等位基因频率≤0.01的VOs(变异观察)。图2和表1分别详细展示了工作流程各阶段被过滤的VOs数量及比例。后果过滤器首先移除了35%具有VEP注释影响的VOs;随后,剔除在gnomAD数据库中呈纯合状态的VOs,使剩余VOs减少27%。合子性过滤器的有效性因遗传模式而异:对于AD关联,11%的剩余VOs被过滤;XLR关联为51%;而AR疾病-基因关联高达94%。对于早发性AD条件,通过合子性过滤的VOs中有68%因存在于gnomAD数据库而被剔除。此外,AD疾病-基因关联的VOs中79%、XLR关联的83%以及AR关联的74%因在未患病参与者中呈现预期合子性状态而被移除。最终,基于OSMP提供数据的所有过滤步骤后,剩余305个VOs涉及226名参与者。在所有遗传模式中,81%的剩余VOs(n=248)因与OMIM疾病概要表型重叠不足被过滤。通过人工审查OSMP外部数据源,46%的剩余VOs(n=26)被进一步剔除,剔除依据包括ClinVar分类为良性/可能良性、家族成员中未适当分离变异,以及外部临床记录显示与OMIM疾病表型重叠不足。经过全部过滤步骤,最终保留31个VOs(占原始查询结果的0.24%),涉及20个新描述的疾病-基因关联和25名参与者。这些VOs被优先提交多学科团队审核:其中70%(14/20)为AD疾病关联,30%(6/20)为AR疾病关联。截至目前,已有1例确诊。在POLR3B基因中发现一个先前未识别的VOs,该患者具有与近期描述的脱髓鞘性Charcot-Marie-Tooth病1I型(OMIM 619742)相似的神经系统特征。该疾病-基因关联在参与者最后一次GWS分析6个月后才被报道,因此当时未被优先考虑。后续临床诊断实验室确认该POLR3B变异为新生变异,并分类为可能致病。本次测试表明,OSMP能够并行查询大量数据集,检索大量VOs及个体表型信息,并以支持查询者根据假设应用自定义过滤工作流程的格式展示这些VOs。
第二次单向匹配测试查询了一组新型候选基因(命名为新型候选基因测试)。这些候选基因是通过分析Care4Rare研究计划中未解决的RGD患者全基因组测序数据识别得出的(筛选标准详见Osmond等人文献)。其中大部分参与者已在Genomics4RD数据库中有记录,可通过OSMP进行查询。本次测试有两个目标:首先,验证单向匹配工作流程的有效性(即我们能否识别出携带这些基因中强候选变异的真实阳性参与者);其次,发现具有相同基因罕见变异和重叠表型的额外家庭,从而为新型疾病-基因关联提供证据支持。本次测试共查询了178个先前识别的新型候选基因,这些基因的大小均在OSMP内存阈值范围内。在178个候选基因中,有140个基因在Care4Rare家庭中曾被优先确定为致病性或强候选变异(即真实阳性),且这些变异可通过OSMP返回(即它们为单核苷酸变异/小插入缺失,且家族数据存在于Genomics4RD中),因此该组基因被用于验证我们的单向匹配工作流程。
上述140个携带真实阳性VOs的新型候选基因通过OSMP进行查询。应用所有过滤器后,89%(124/140)的候选基因的VOs仍保留在我们的优先列表中。在124名携带通过工作流程过滤的真实阳性VOs的参与者中,仅有一名参与者的另一个新型候选基因的VOs也通过了全部过滤流程。这些案例已按后续章节所述提交多学科团队审核。对于16个未通过全部过滤器的基因:其中6个基因的VOs被后果过滤器移除,主要因变异发生在经典剪接位点附近;9个基因的VOs被gnomAD杂合过滤器剔除,因这些基因与严重AD儿童期发病相关,但变异存在于至少一名gnomAD个体中。所有这些案例中,gnomAD中的变异先前已被识别,并被认为是弱新型候选变异。最后,1个基因的VOs被患病状态过滤器移除,因变异存在于标记为未患病的家族成员中;但经复核后,该亲属的患病状态被判定为不确定。我们认为这些假阴性结果无需调整当前单向匹配协议。
在针对178个新型候选基因的OSMP查询中,我们排除了上述携带真实阳性变异的家庭,以寻找具有相同新型RGD表型的其他家庭。OSMP共返回了20,308个等位基因频率≤0.01的VOs。图3和表2分别详细展示了工作流程各阶段被过滤的VOs数量及比例。总体而言,后果过滤器移除了28%符合条件的VOs,gnomAD杂合过滤器则剔除了25%的剩余VOs。与OMIM基因测试类似,合子性过滤器的有效性因这些新型候选基因的假设遗传模式而异:约3%的AD基因VOs、65%的XLR基因VOs和91%的AR基因VOs被该过滤器移除。对于疑似早发性AD条件的基因,剔除gnomAD中存在的变异使剩余VOs减少了75%。在过滤未患病参与者中的VOs时,82%的AD疾病-基因关联VOs、45%的XLR关联VOs和65%的AR关联VOs被移除。基于OSMP提供数据的所有过滤步骤后,剩余604个VOs。通过过滤表型重叠不足的参与者,所有遗传模式下的VOs被移除了91%。最后,约67%的剩余VOs通过OSMP无法直接获取的外部数据被排除。用于排除这些VOs的外部数据与OMIM基因测试类似,包括未纳入Genomics4RD的家庭成员测序数据、更详细的临床表现记录以及XLR基因在gnomAD中的半合子变异携带者数量。经过全部过滤步骤后,最终18个VOs(占初始查询结果的0.09%)涉及14个新型候选基因,被提交多学科团队审核。其中10个基因疑似与AD相关(10个杂合VOs),3个基因疑似与AR相关(2个纯合VOs和同一参与者的2个杂合VOs),1个基因疑似与XL条件相关(1个半合子VOs)。这些新型候选基因中优先VOs的审核工作仍在进行中。
讨论
OSMP测试版的开发与测试表明,单边匹配机制能有效识别RGD表型未确诊患者中具有临床意义的遗传变异。尽管每次测试返回数万个VOs,我们建立了高效工作流程可快速筛选出最可能致病的变异。在新发现的OMIM基因POLR3B中已确诊1例。剩余优先VOs将由多学科团队(含患者临床医生)审核,预计通过临床实验室验证(针对已知致病基因)、功能实验(评估VO对基因功能的影响)及提交至数据共享计划(如MME)等后续工作,将在更多基因中实现确诊。
OMIM基因及新候选基因测试表明,在进行此类匹配时提供受试者个体信息对最大限度排除假阳性至关重要,以在开展全面病例审查前完成初步筛选。了解变异在未患病受试者中的存在情况,能高效过滤所有遗传模式相关基因的VOs,分别使OMIM基因测试和新候选基因测试中剩余VOs减少78%和79%。同样,以HPO术语形式获取患病受试者的表型数据,使单边匹配测试中剩余VOs的排除率超过80%。此外,VO的合子信息对已知或假设为常隐遗传(AR)模式的基因尤为有效,使两项测试研究中剩余VOs的排除率均超90%。表型与基因型数据在主动排除潜在匹配中的效用,本团队在双边匹配中亦有类似经验——当初始匹配时提供此类信息,超半数双边匹配被排除。提高MME提交匹配中此类个体信息的纳入率,已被视为提升双边匹配效率的关键因素,而我们的测试表明,若无此类数据支持,单边匹配平台将难以成功。
用于这些测试的单边匹配工作流程通过检查Care4Rare在新候选基因中先前优先的VOs是否通过所有过滤器而得到验证。约89%的这些真阳性VOs通过了所有过滤器。对16个未通过所有过滤器的真阳性VOs的检查突出了未来基因查询调整的机会。16个真阳性VOs中有6个被后果过滤器移除,因为它们位于内含子中,且根据SpliceAI预测不会影响剪接。可以考虑放宽后果过滤器以包括所有内含子变异,从而提高工作流程的敏感性;然而,这将导致在后续过滤步骤中需要审查的VOs数量显著增加。同样,16个真阳性VOs中有9个被gnomAD杂合过滤器过滤掉。如果放宽此过滤器以包括任何gnomAD等位基因频率为0.0001或更低的VO,所有九个VOs将通过剩余过滤器。然而,当尝试这样做时,四倍多的VOs需要在表型过滤器处进行审查。我们建议,虽然gnomAD杂合过滤器可以在未来的查询中放宽以提高敏感性,但这一决定应权衡诸如表型严重程度和用于手动审查VOs的人力资源等因素。
结论
OSMP测试版可实现基因层面的单边匹配查询,旨在优先筛选RGD表型未确诊患者中具有临床意义的变异。针对新发现疾病基因关联及新候选基因的单边匹配工作流程开发与测试,既揭示了基因查询返回的VOs数量规模,也凸显了变异层面和受试者个体数据在筛选合理可审匹配量中的重要性。我们的测试验证了单边匹配能发现新候选基因中更多确诊案例和意义变异的可行性。通过测试OSMP测试版获得的经验将用于优化平台功能,这些洞见对开发同类工具的团队具有参考价值。将更多RGD数据库接入OSMP等单边匹配服务,对扩大临床未确诊RGD家庭的匹配覆盖面至关重要,最终目标在于明确其疾病的遗传病因。


















