脊髓性肌萎缩(Spinal Muscular Atrophy, SMA)是一种常染色体隐性遗传病,主要影响婴儿和儿童。大多数SMA患者的SMN1基因发生了突变,导致他们体内缺乏一种叫做运动神经元存活蛋白(SMN蛋白)的重要蛋白质。这种蛋白质对于运动神经元的生存至关重要,缺乏它会导致肌肉无力和萎缩。(参见《异常的外显子跳跃与脊髓性肌萎缩》一文)
到目前为止,全球有3款SMA疗法获批,《诺西那生纳如何治疗SMA》一文已经详细介绍了基于反义寡核苷酸药物(Antisense Oligonucleotides, ASO)的治疗方法,本文则聚焦曾被称为“史上最贵药物”的基因疗法Zolgensma,看它是如何帮助SMA患者的。
基因治疗的基本原理
近些年来,基因治疗及基因编辑疗法成为现代医学中重要的治疗手段。以Zolgensma为代表的基因替代治疗(Gene Replacement Therapy)通过将正常或修饰的基因导入患者体内,以纠正或替代体内缺陷基因的功能,从而达到治疗效果,此类疗法一般针对已知致病基因的单基因遗传病。
基因递送载体分为病毒载体和非病毒载体,前者包括腺病毒(Adeno virus, AdV)、腺病毒相关病毒(Adeno-associated virus, AAV)和慢病毒(Lenti virus, LV),以其高效的感染能力著称。AAV由于其低致病性和低免疫原性,并且不会整合到宿主基因组中,减少了潜在的插入突变风险,而被认为更安全,得到了广泛使用。
野生型AAV具有一个二十面体的蛋白质外壳和一条长为4700碱基对的单链线性DNA,AAV基因组包括两个主要开放阅读框:Rep基因和Cap基因,以及两端完全相同的末端反向重复序列(inverted terminal repeats, ITR)。重组AAV(recombinant AAV, rAAV)保留了对病毒复制和包装起关键作用的两端ITR,而将AAV包含的其余基因代替成为治疗基因。这样,rAAV既保留了野生型AAV可以在细胞中形成染色体外环状DNA的特质,又可以持续稳定地表达治疗基因。
Zolgensma正是利用了rAAV作为基因治疗的递送工具。因此,不同于需要终身用药的诺西那生纳(Nusinersen)和利司扑兰(Risdiplam),Zolgensma只需一次静脉注射便可提供长期治疗效果。
递送载体AAV的前世今生
虽然早在1960年代,AAV就作为腺病毒的污染物被发现,但当时的研究重点关注将腺病毒改造成基因治疗载体。然而,1999年9月17日,年仅18岁的患者Jesse Gelsinger在接受腺病毒载体基因治疗临床试验后不幸去世。后续研究发现,Gelsinger的死亡是由于他对腺病毒载体产生了严重的先天免疫反应,导致多器官衰竭。这一事件引发了科学界对基因治疗安全性的广泛关注,促使科学家寻找更安全的基因递送载体。
到了本世纪初,科学家逐渐意识到AAV可以作为一种安全有效的基因递送工具,宾夕法尼亚大学的James M. Wilson团队在这方面做出了重要贡献。当时在Wilson实验室担任研究员的高光坪(现任麻省大学医学院李伟波罕见病研究中心主任、红瑞基因治疗中心主任、教授,也是瑞鸥科学顾问委员会成员)在研究中首先分离出了AAV7和AAV8血清型,这些AAV的基因递送效率比当时已知的载体高出10-100倍。
随后,在2003年,高光坪和Wilson分离出AAV9,这种血清型拥有更高的基因递送效率。接着,俄亥俄州Nationalwide儿童医院的Brian K. Kaspar团队发现AAV9能通过静脉注射,穿过通常阻挡药物和病毒进入大脑的保护屏障——血脑屏障。正是这一发现使得Zolgensma采用AAV9作为载体,可以通过静脉注射在中枢神经系统中广泛表达。在两位接受过Zolgensma治疗的患者组织样品中,其大脑、骨骼肌、心脏和肝脏都检测到了SMN蛋白的表达。
除此之外,还可以通过直接向脑脊液注射的方式递送药物至中枢神经系统,如脑室内(intracerebroventrivular, ICV)注射和鞘内(intrathecal, IT)注射。目前,Zolgensma的鞘内注射临床试验仍在进行中。
Zolgensma的作用机制
2019年5月,Zolgensma(通用名onasemnogene abeparvovec)通过美国FDA批准,它是由诺华制药(Novartis)收购基因治疗公司AveXis后研发的治疗SMA的药物。作为一种基因疗法,它被批准应用于2岁以下,携带SMN1双等位基因突变的SMA患者。
截至目前,Zolgensma已经在全球超过51个国家批准,帮助了超过3700名患者。中国也于2022年开始了Zolgensma的相关临床试验(药物名称:OAV101注射液,药物临床试验登记与信息公示平台,CTR20220992;CTR20240883)。
上面已经提到,Zolgensma是一种采用AAV作为载体的基因疗法。它通过一次性注射,将健康的SMN1基因送入患者体内,帮助他们重新生成SMN蛋白。这种方法可以有效地阻止运动神经元的进行性丢失,从而改善患者的肌肉功能(图1)。
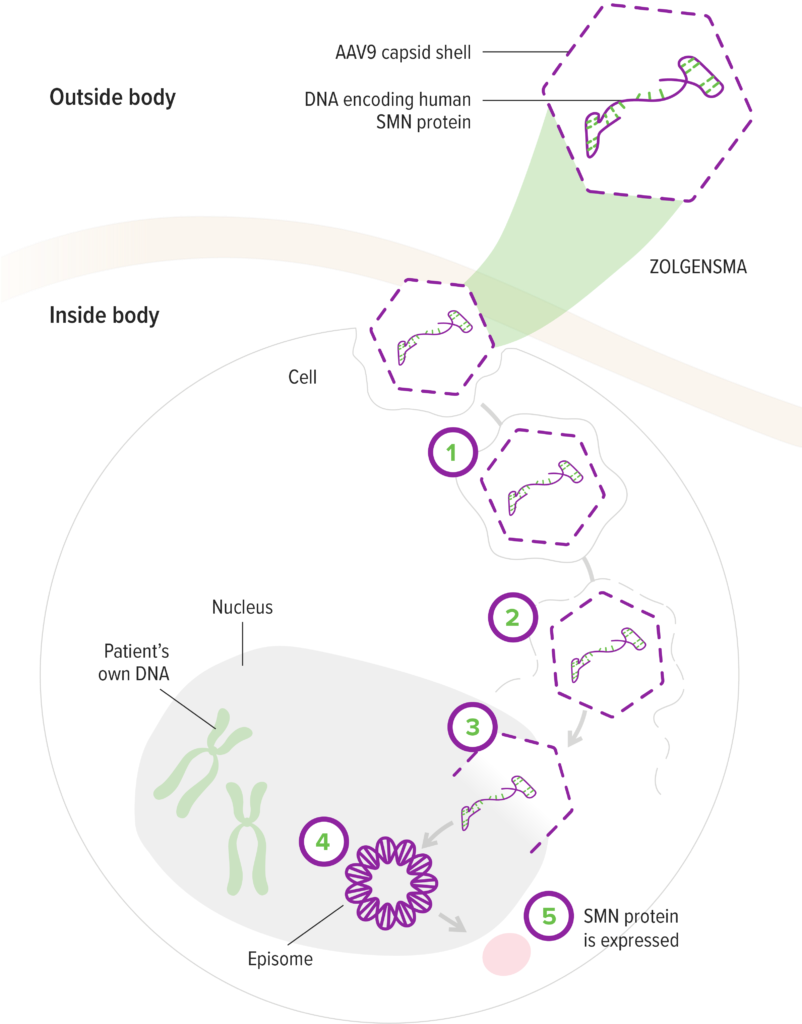
▲图1:Zolgensma的作用机制。① AAV9载体进入运动神经元 ② AAV9载体将SMN基因递送至细胞核 ③ SMN基因在靶细胞中形成重组互补DNA片段 ④ 在停止分裂的运动神经元中,形成染色体外环状DNA ⑤ SMN蛋白快速持续地在运动神经元中表达(来自https://www.zolgensma-hcp.com/)
Zolgensma的临床试验进展
目前,Zolgensma已在美国及其他多个国家开展临床试验并批准上市,显示了在治疗SMA患者方面巨大的潜力和实际应用效果。
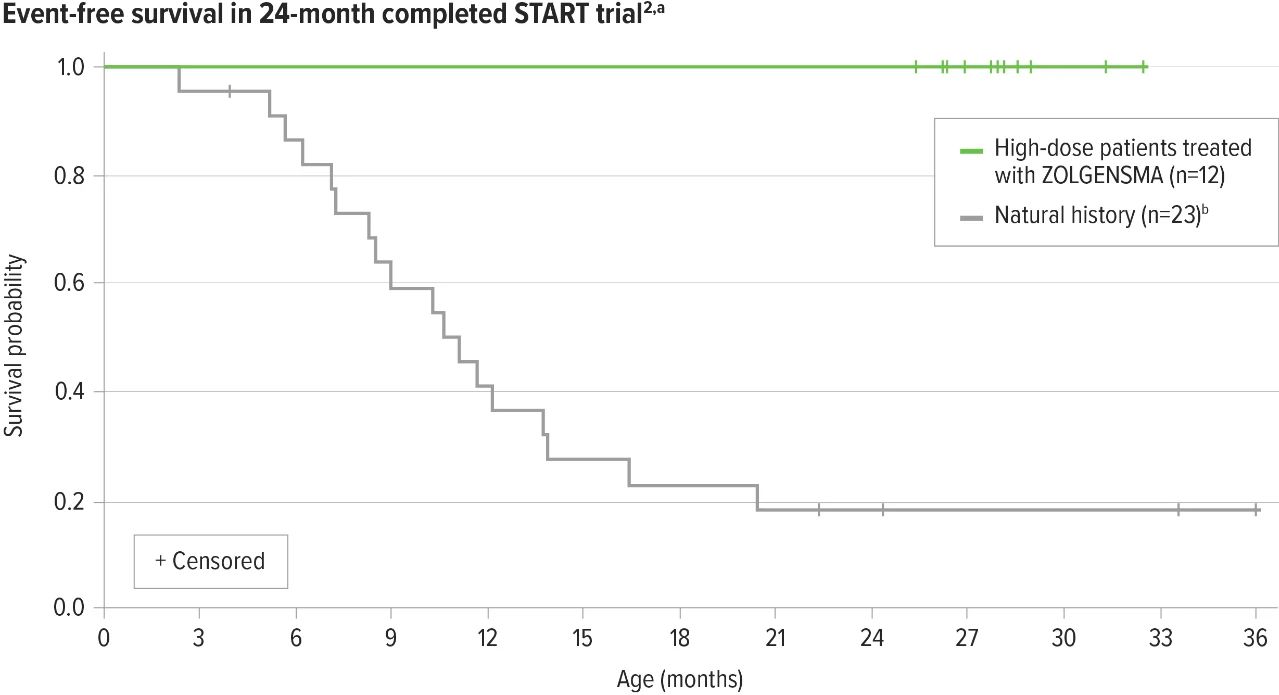
▲图2:START临床试验的24个月无事件生存期
Zolgensma的局限性
虽然Zolgensma有很大的潜力,但它也存在一些局限性。
首先,目前它只被批准用于2岁以下的SMA患者。对于超过2岁的儿童患者以及慢性SMA患者的疗效还有待临床试验的结果。中国也参与了针对超过2岁,小于18岁,初治的2型SMA患者鞘内注射Zolgensma的疗效和安全性研究(药物临床试验登记与信息公示平台,CTR20220992)。
其次,Zolgensma是基于AAV9的基因疗法。由于AAV遍布于自然界,患者若从母体或自然界接触过AAV9并形成AAV9中和抗体,则不适用于此类疗法。这是因为体内的中和抗体会排斥AAV9载体,造成身体的免疫反应,抑制其携带的SMN基因表达。因此,准备接受Zolgensma治疗的患者必须首先接受AAV9中和抗体的检测,不超过标准1:50及以下方可进行治疗。
此外,Zolgensma也被报道会激发急性肝损伤,急性肝衰竭和谷丙转氨酶升高。因此,接受Zolgensma治疗的患者需要在治疗前后接受肝功能检查、肌酐检查和全血细胞计数。使用皮质类固醇药物可以帮助减轻这种副作用。
最后,如本文开头就提到的,作为“史上最贵药物”,Zolgensma的高成本也是一大挑战,每次治疗费用高达200万美元,这对许多家庭来说是巨大的经济负担。
结语
除Zolgensma之外,美国FDA批准了其他基于AAV载体的基因治疗方法,如由Spark Therapeutics开发的用于治疗视网膜疾病的Luxturna,由Serepta Therapeutics开发的用于治疗杜氏肌营养不良(Duchenne Muscular Dystrophy, DMD)的Elevidys,以及用于治疗血友病的Hemgenix和Roctavian,这些疗法都展示了基因治疗的巨大潜力。
Zolgensma为SMA患者带来了新的希望,通过一次性静脉注射即可提供长期的治疗效果。未来,随着研究的不断深入,基因治疗和基因编辑疗法将发挥越来越重要的作用,为更多的罕见遗传疾病患者提供有效的治疗方案,为全人类的健康事业书写全新的篇章。


















