《患者参与药物监管决策的路径研究》
在2024国际罕见病日中国区宣传周期间,由蔻德罕见病中心联合中国药科大学国际医药商学院茅宁莹教授团队开展的《患者参与药物监管决策的路径研究》报告重磅发布!报告旨在摸清目前各方对患者参与的认知及需求,让患者参与药物监管的路径和模式更加明晰,并建立良好的、规范化的患者参与机制,提升患者在药物监管过程中的参与广度与深度。
“罕见病信息网”将独家展开全文连载,敬请读者关注和转发。本文为连载第六篇:《患者参与药物监管的国际经验:美国患者参与药物监管体系建设》。
第一篇:《患者参与药物监管的相关概念界》
第二篇:《患者参与药物监管的动力》
第三篇:《企业及其他相关方对患者参与药物监管有哪些需求?》
第四篇:《患者参与药物监管的理论基础》
第五篇:《WHO患者参与药物监管的经验》
美国患者参与药物监管体系建设
概念界定
依据MedicineNet中的词条解释,“患者”通常被定义为“正在接受由有执照的医学治疗从业者指导的所需专业服务,以维持、改善或保护健康或减轻疾病、残疾或疼痛的个人”;据美国陆军医疗司令部的定义,也包含接受受过医学训练的人员的医疗护理或治疗的生病、受伤或受伤的士兵。
在西方国家,患者组织(Patient Organization/ Patient Groups)是非营利组织,一般来说,是由某一种或一类疾病的患者或家属发起成立,代表和维护该疾病患者社群整体利益,为患者家庭提供支持与服务的社会团体。这些组织倡导患者的权利、提供信息和资源、资助研究和临床试验[]。如美国癌症协会、全国精神疾病联盟和美国心脏协会等,这些组织通过外展、会议、咨询、网站和出版材料,倡导并为数百万患有癌症、精神疾病、糖尿病和心血管疾病等身心疾病的人提供服务。
患者参与历史沿革
PART/1 公众参与基础
自1776年美国独立后,由建国初期的邦联制政体转为联邦制,建立了联邦政府,并在1787年举行制宪会议,确立了三权分立以保障公民权益。同时,美国宪法和法律条文也规定了对公民言论、集会、选举等权利的保护。
在民主原则的环境下,美国公众参与日渐成熟。依据参与主体组织化程度的不同,可将各主要参与方式分为个体参与和组织参与。前者主要包括选举投票参与和官民个别接触,后者主要包括通过政党和政治团体参与。在美国,公民可以通过上述两种方式表达自己的利益诉求,参与和影响公共政策的制定。
PART/2 患者参与发展历程
自1988年美国成立艾滋病协调办公室以来,FDA开始逐步重视患者在监管和决策中发挥的作用。从最初的患者代表发展到患者参与协作组织,FDA不断扩大患者参与药物监管的群体、拓宽患者参与监管的路径并规范患者参与行为,并形成了以患者为中心的药物开发计划(PFDD),将患者作为FDA的工作核心(如图3-1)。

图3-1 FDA患者参与过程演变
顶层设计规划
PART/1 监管机构部门设置
自1988年以来,FDA实施了许多新的举措来促进患者的参与,包括将患者感受报告纳入数据审查中、提高患者的发言参与机会等。为了直接与患者或患者社群组织合作,并鼓励和支持他们积极参与FDA的决策和政策制定,FDA设立了卫生与组织事务办公室(OHCA)。该部门作为负责患者组织的窗口部门,通过招募新的患者代表、召开患者代表研讨会或网络研讨会等方式,开展系列培训活动以让患者更好地理解药事相关法律法规。
FDA在2014年建立了患者联络网站,旨在促进患者参与,加强OHCA的培训与宣传活动,也为患者、护理人员和患者社群组织提供信息及交流的平台。
2016年FDA成立了安全创新法案(FDASIA)1137工作组,目前已发展成为跨中心的患者委员会。该委员会旨在将所有患者中心和办公室汇集在一起,在监管决策中和整个产品生命周期中更好地协调和整合患者意见。
此外,FDA还在下属的专员办公室之一的临床政策和计划办公室(Office of Clinical Policy and Programs,OCPP)中设置了患者事务专员(Patient Affairs Staff),致力于为患者社区提供与FDA互动的机会和平台,并通过各种举措扩大公众的参与意识,帮助患者、护理人员和倡导者把握参与监管的流程和机会。
PART/2 相关政策制度体系
2012年安全创新法案(Food and Drug Administration Safety and Innovation Act,FDASIA)中提出了患者参与医药产品讨论,并加强了FDA保障和促进患者公共健康的能力,认可了患者意见的价值并促进患者更早地参与医疗产品审查的监管过程。
2012年,FDA建立了聚焦患者的药物研发(Patient-Focused Drug Development,PFDD)计划,以便更系统地了解患者对特定疾病及其当前可用治疗的看法。
2012年到2017年,基于《处方药使用者付费法案V》(Prescription Drugs User Fee Act V,PDUFA V),FDA召开了24次针对特定疾病患者的PFDD会议,例如斑秃、慢性疼痛、亨廷顿病、帕金森病、肺动脉高压等。
2015年,CTTI实施了Enhance Patient Engagement项目,通过对患者组织、企业及学术机构的研究人员的采访等,完成了患者组织参与药物研发的建议报告。
2016年,在新颁布的《21世纪治愈法案》(21st Century Cures Act)支持下,PFDD成为FDA的一项优先事项。
2019年3月,FDA和CTTI基于《处方药使用者付费法案Ⅵ》举办了主题为“加强患者观点纳入临床试验”的公开研讨会,目的是加强患者对临床试验访问、设计、实施和试验后随访的看法。该研讨会收集了患者、护理人员、行业、学术研究人员和专家从业者关于患者参与和保留临床试验的挑战和障碍的意见。
2021年6月,FDA发布了在癌症临床试验评价中使用从患者或受试者直接获得的信息(Patient Report Outcome,PRO)的指导原则草案。
截止2023年5月,FDA已经发布了PFDD指导原则1、指导原则2、指导原则3草案和指导原则4草案。
患者参与药物监管的实践模式
PART/1 患者参与相关计划
(1)以患者为中心的药物开发(PFDD)
FDA认识到通过PFDD会议收集患者意见的价值,患者意见也是FDA药品获益风险评价和审评决策的重要因素之一。每次PFDD会议后,FDA会在患者报告中总结患者和患者代表的意见,在FDA官网公开每次会议的详细日程和讨论内容等信息。
① 由FDA或主导的PFDD[45]:
以患者为中心的药物开发(PFDD)是一种系统性方法,有助于确保患者的经验、观点、需求和优先事项被捕获并有意义地纳入药物开发和评估(如图3-2)。患者作为与病情共存的专家具有独特的优势,可以为药物开发和评估的治疗环境提供信息。FDA工作人员负责领导计划,并在药物评估和研究中心(CDER)内提供战略、监管、计划和政策援助,以促进将患者意见纳入决策。

图3-2 FDA以患者为中心的药物研发路径图
② 由外部主导的PFDD:
为了帮助扩大FDA的PFDD计划的益处,FDA也欢迎患者组织主导拟定和组织以患者为中心的合作,以其他疾病领域产生公众意见,使用FDA领导的PFDD会议建立的流程作为模型。
(2)FDA患者倾听会议
FDA患者事务部(FDA Patient Affairs)自2018年开始举办患者倾听会议。这些会议是医疗产品中心与患者及其倡导者互动的渠道。患者倾听会议是患者和倡导社区通过直接与FDA工作人员交谈来分享经验和观点的众多方式之一。该会议只有FDA、患者、护理人员、倡导者和社区代表可以参加。
患者倾听会议可以是FDA要求的(FDA有一组特定的问题需要征询患者意见)或患者主导的(当患者社区希望与FDA分享他们的观点时)。
患者倾听会议有助于FDA为医疗产品开发、临床试验设计、患者偏好提供信息,并塑造其监管思维。在患者倾听会议期间,FDA工作人员将提出问题或只是倾听,以更好地了解患者的经验和观点。
(3)患者参与协作组织(PEC)
2017年,患者社区在公众反馈中发表评论,以创建一个外部小组,为整个机构的患者参与提供意见。为了响应这些反馈,在患者组织代表的帮助下,FDA和临床试验转型计划(CTTI)以EMA的患者和消费者工作组为蓝本,建立了患者参与协作组织(PEC)。PEC由联邦法律保障,以促进患者参与并将患者体验纳入监管过程。
患者参与协作组织(PEC)中包含患者组织和个人代表,他们讨论如何在FDA的医疗产品开发和其他监管讨论中实现更有意义的患者参与。
(4)患者参与咨询委员会
患者参与咨询委员会主要职责为在每年举办的公共咨询委员会会议上,就与医疗设备、设备监管以及患者使用医疗设备有关的复杂问题向专员或指定人员提供建议。委员会由九名有表决权的成员、一名无表决权的行业代表和一名有表决权的消费者代表组成。对于需要核心成员以外的专业知识的特定会议,委员会可以联系参加FDA和美国国立卫生研究院的该小组和其他咨询小组的专家。
(5)患者代表计划
患者代表计划是FDA的旗舰计划。FDA为患者和护理人员提供机会,在FDA监管医疗产品(药物、生物制剂和设备)时向其提供关键建议。其具体实施路径在下一小节详述。
PART/2 患者参与药物监管典型模式——患者代表计划
FDA患者代表计划和FDA患者代表是美国卫生与公众服务部的服务标志。
(1)FDA患者代表的角色
在FDA患者代表计划中,患者和倡导者以临时雇员的形式被任命为特殊政府雇员(Special Government Employees,SGE),直接为FDA工作人员提供他们对各种疾病、病症和医疗设备的经验与见解,并可以访问机密信息。FDA任命所有咨询委员会成员(行业代表除外)为SGE。
FDA患者代表在参与FDA咨询委员会协商时,能够与科学成员和其他专家互动。
代表候选人经过精心招聘和培训,为机构主办的各种会议和活动做好准备。通过这种参与,进一步帮助FDA了解患者的需求、优先事项和偏好,并收集有意义的数据,为医疗产品开发和决策提供信息。
(2)患者代表招聘领域
FDA根据实际需要招募FDA患者代表,以下是目前招聘的一些领域(如表3-1):
表3-1 FDA患者代表招聘领域
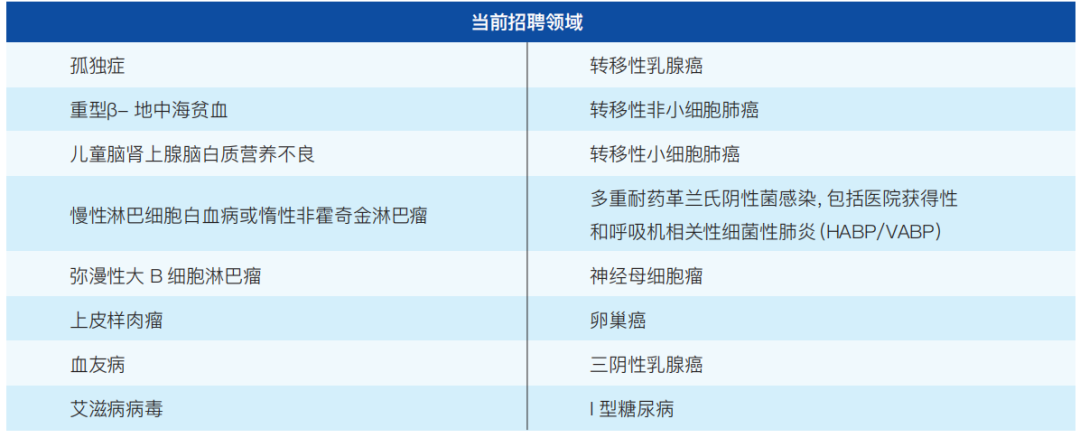
注:根据FDA官网绘制,数据截至2023-12-04
(3)FDA患者代表遴选标准
FDA根据以下标准选择FDA患者代表。除了要求申请人必须是至少18岁的合法美国公民外,还需:
拥有作为患者或主要护理人员的个人疾病经历;
能够客观地代表社区中其他人的观点;
愿意和有能力公开表达他们的观点和意见;
了解大多数治疗方案,并对他们所代表的疾病领域有一定了解;
公平公正,要求本人或近亲成员的几乎没有道德问题或财务利益冲突(例如,可能受FDA决定影响的公司的财务利益,如股票)。
(4)FDA患者代表的主要活动
①在FDA咨询委员会和小组中任职,以获得有关科学、技术和政策事项的独立专家建议。
②通过直接与部门审查人员接触,在监管医疗产品开发和审查过程中尽早向FDA相关机构部门提供意见。
(5)利益冲突
为了担任FDA患者代表,候选人将被任命为特殊政府雇员(称为“SGE”)。一旦被任命,FDA患者代表必须遵守所有联邦政府的利益冲突要求和道德准则。在为FDA机构开展工作之前,所有候选人都要经过利益冲突审查,机构工作人员将协助进行筛选过程。
PART/3 患者参与药物监管具体实践—— CDRH患者科学和参与计划
患者科学和参与计划是由FDA医疗器械和辐射健康中心(Center for Devices and Radiological Health,CDRH)发起,由社会科学家、健康经济学家、统计学家、临床提供者和其他专家组成团队,致力于与患者成为合作伙伴,还负责帮助开发和评估衡量患者体验和观点的工具,包括临床结果的评估、患者偏好信息、患者生成的数据(例如消费者可穿戴设备收集的数据)等,并将患者的观点和相关体验信息纳入医疗设备的开发和评估中,在整个产品生命周期内保障医疗设备的安全有效,进而影响监管决策和行动。
CDRH表示,与患者的合作伙伴关系有助于其推进创新医疗产品的开发和评估,并监督上市医疗设备的性能。且从患者基于科学信息所产生的对临床研究开发的见解中可确定该研究评估中的患者偏好,并确保患者能够从中获得有关医疗设备的相关有用信息,这些信息也有助于医疗保健的决策。
①患者参与的作用:
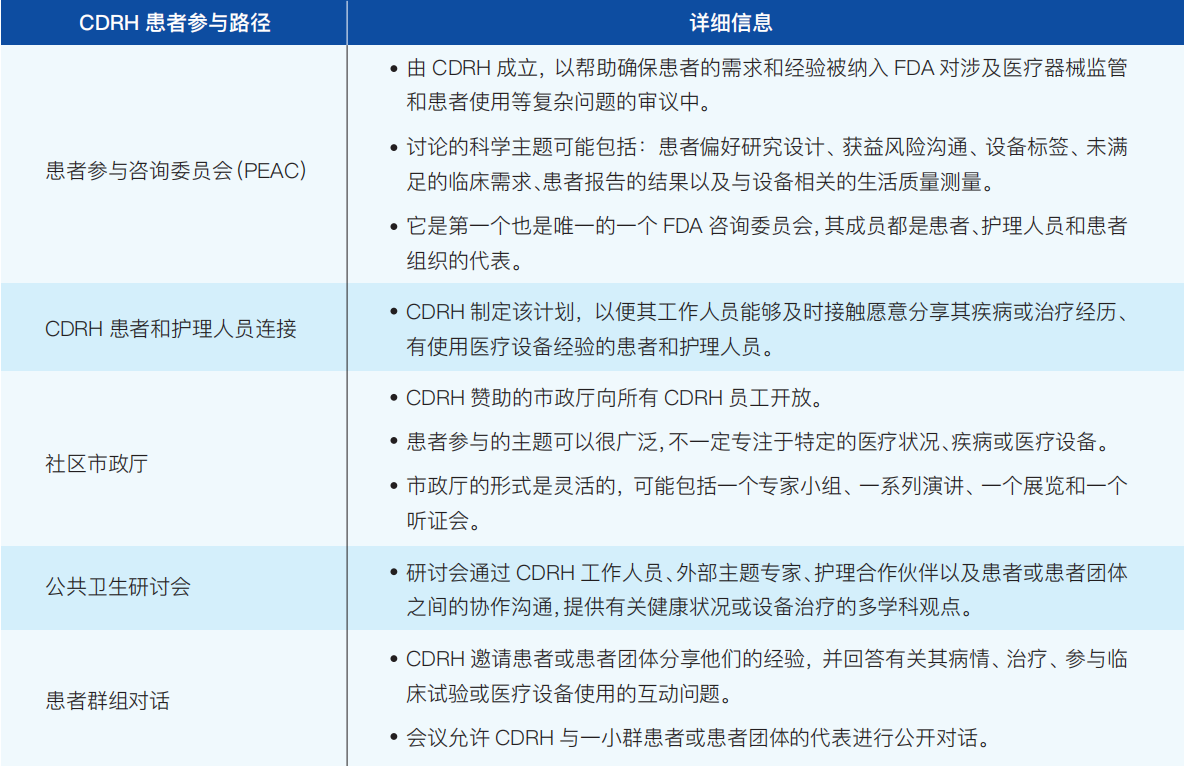
②患者参与的路径(如表3-2):
表3-2 CDRH患者参与路径及详细信息
③患者参与的激励方式:
打通监管部门之间的协作渠道。CDRH与FDA的其他办公室合作,例如患者事务专员办公室和药物评估和研究中心的以患者为中心的药物开发小组。
建立多元化医学创新合作平台。CDRH与医疗器械创新联盟(Medical Device Innovation Consortium,MDIC)外部合作,推进变革式医疗技术发展,促进患者参与临床试验。
鼓励患者与制药企业发展合作伙伴关系。CDRH鼓励医疗器械行业与患者一起设计和进行临床研究。
患者参与药物监管实践成效
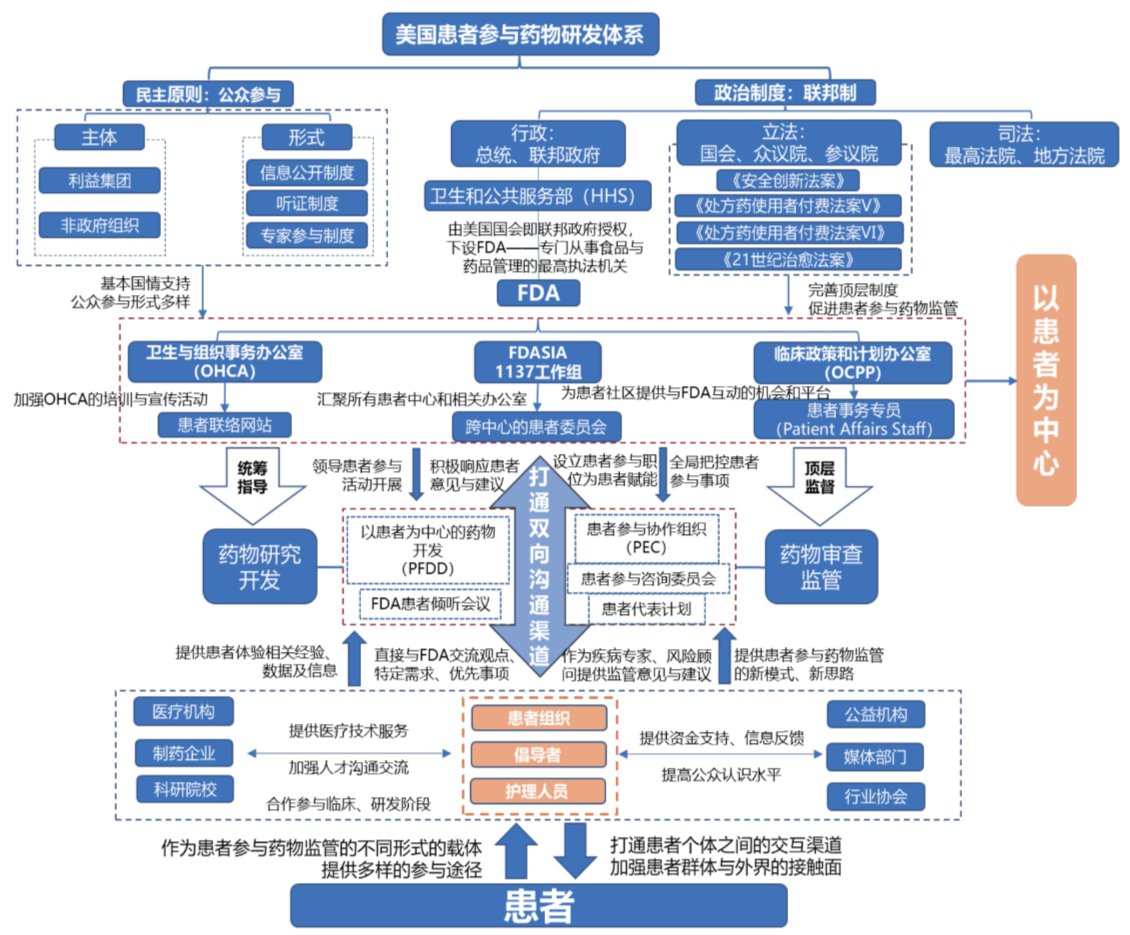
图3-3 美国患者参与药物监管框架图
PART/1 FDA积极回应患者参与咨询委员会会议反馈
PEAC由FDA成立,以确保将患者的需求和经验纳入FDA对涉及医疗器械监管和患者使用医疗器械的复杂问题的审议中。
自2017年以来,PECA陆续召开了以“患者参与医疗器械临床试验”“患者生成的健康数据”、“医疗设备中的网络安全”、“医疗设备中的人工智能(AI)和机器学习(ML)”、“医疗设备召回:以患者为中心的沟通”和“增强现实(AR)和虚拟现实(VR)医疗设备”主题的会议,并在2022年的会议中讨论如何将患者观点纳入FDA和行业利益风险决策,以及与使用或处方该技术相关的医疗保健提供者决策过程。
FDA则根据以上会议反馈采取数项行动,包括(如表3-3):
表3-3 FDA近年对PEAC会议反馈所采取的行动(2018-2021年)

PART/2 特定会议报告提供了大量患者体验相关信息
截至2023年2月,FDA在其官方网页上提供了156份与患者体验数据相关的某些公开外部报告和资源的链接,以供患者社区、患者倡导者、研究人员、药物开发商和联邦机构参考使用。报告包括患者体验相关信息以及与患者体验数据、自然史研究或其他特定状况背景和未满足医疗需求讨论有关的拟议指南草案。
这些特定会议包含了FDA领导的PFDD会议、外部主导的PFDD会议和患者倾听会议,疾病范围囊括了肢端肥大症、急性卟啉病、肾上腺脊髓神经病、斑秃、乳腺癌、克拉贝病等多种疾病,时间跨度从2013年到2023年十年之久,会议主持方也包含了FDA、制药企业、基金会和国家罕见病组织等。
PART/3 患者偏好信息(PPI)被纳入医疗器械决策
患者偏好信息可以为医疗设备的设计提供信息、影响临床研究的设计方式,并能用于了解临床研究结果对患者的影响,例如:
在医疗器械开发的早期识别患者未满足的需求
从患者的角度确定技术最重要的获益和风险
评估临床研究结果对患者的重要性
确定研究结果的有意义的变化
阐明患者如何看待给定技术的收益和风险的权衡
显示不同群体患者对获益和风险的偏好如何变化,以及他们接受不确定性的意愿
PPI已在多个药物申报中使用,并为CDRH的监管决策提供了信息。目前,由行业赞助的23项患者偏好研究已经完成或正在进行中。
PART/4 患者组织参与政府机构意见征询
患者组织通常在教育公众和游说政府官员方面发挥重要作用,目的是增加研究和治疗的研究经费以及改变与他们所代表的疾病相关的立法。他们游说国家研究所增加支出卫生部(NIH)和美国国防部。一些组织甚至在经济上支持自己的研究。例如,美国癌症协会在2011年的收入接近10亿美元,赞助了重要的临床研究和肿瘤学临床医生的继续教育,并赞助了许多其他地方和国家活动。
在过去的几十年里,其中许多团体实现了他们的目标。原因之一是许多PAO是由患有严重疾病的患者或前患者创立和经营的,它们在公众、立法者和政府中具有可信度。政府机构(如NIH)因此经常被征求意见,例如,PAO代表参加NIH委员会,审查研究提案并参加国会听证会。
经典案例
(1)患者参与助力镰状细胞病(SCD)基因疗法研发
镰状细胞病(SCD)由β-珠蛋白(构成血红蛋白的一种蛋白质)的特定基因突变引起,其降低了血红蛋白的溶解性并增加了红细胞的不稳定性,因此,患者体内存在着大量镰刀状的异形红细胞。大部分患者会因疾病的反复发作而逐渐失去脾功能,出现免疫力低下、全身多器官并发症,严重影响生活质量,甚至猝死。
2023年12月8日,美国食品和药物管理局(FDA)批准了首款基于细胞的基因疗法Casgevy和Lyfgenia,用于治疗12岁及以上患者的镰状细胞病(SCD)。两种治疗方法均根据单独的临床试验获得批准,接受治疗的患者显示出显着的改善。标志着基因治疗领域的里程碑式进步。
在美国FDA批准这两种镰状细胞病基因疗法的过程中,患者以及研究人员、临床医生和倡导团体在推动这些基因疗法的开发和批准方面发挥重要作用。在药物研发过程中,患者不仅参与接受长期研究的跟踪,以评估每种疗法的长期安全性和有效性,还在接受治疗的过程中与研究人员分享患病及治疗经历。据美联社报道,接受基因治疗的维多利亚·格雷在一次科学会议上与研究人员分享了她的患病及参与基因治疗试验的经历。
在FDA有关这两种疗法的审批相关的咨询委员会中,公司发布的一份简报文件中表示,46人在这项关键研究中接受了治疗。在接受了至少18个月随访的30人中,有29人至少一年没有出现疼痛危机,而且所有30人在这么长时间内都避免了因疼痛危机住院。在咨询委员会的成员中,除了有FDA外部专家,同时也有患者和患者倡导者担任委员会的成员,在讨论和决策过程中提供他们独特的观点。
镰状细胞病基因疗法的开发过程中,患者的积极参与发挥了重要作用,除了参与临床试验,提供关于治疗效果和安全性的宝贵反馈,患者的经历和简介对于制定治疗政策和决策产生了重要的影响,有助于确保治疗方案符合患者的需求,发挥着不可或缺的作用。各利益相关者之间的共同努力共同促进公众健康和科学的发展。
(2)患者意见助力法布雷病治疗药物的审批
法布雷病(Fabry disease)是一种罕见的X连锁遗传性疾病,由于X染色体长臂中段编码α-半乳糖苷酶 A(α-Gal A)的基因突变,导致α-半乳糖苷酶A结构和功能异常,使其代谢底物三已糖神经酰胺(Globotriaosylceramide,GL-3)和相关鞘糖脂在全身多个器官内大量堆积所导致的临床综合征[]。法布雷病常表现出皮肤、眼、耳、心脏、肾脏、神经系统及胃肠道等症状。
在聆听法布雷疾病的会议中,患者和护理人员表明酶替代疗法(ERT)可改善肾脏功能并有助于缓解疲劳,并且胃肠道症状是法布里患者和护理人员在会议上提到的最常见症状,但是ERT常常没有明显改善。
酶替代疗法,是利用基因重组技术体外合成α-Gal A替代体内缺陷的酶的治疗方法。2003年,Genzyme公司研发的阿加糖酶β(商品名Fabrazyme),由于改善法布雷病患者的肾功能效果显著,获得美国FDA加速批准,成为首个用于治疗年龄≥2岁、确诊为法布雷病的成人和儿童患者的药物。2023年5月9日,FDA批准了Chiesi Group与Protalix BioTherapeutics合作开发的水解溶酶体中性糖鞘脂特异性酶(聚乙二醇化酶替代疗法),商品名为Elfabrio。研究数据表明Elfabrio的安全性、耐受性和有效性在140多名患者(未接受过ERT治疗的患者和接受过ERT治疗)中进行了长达7.5年的随访治疗研究,包括一项达到主要终点的头对头试验。临床结果证明Elfabrio在控制估计肾小球滤过率 (eGFR) 下降方面的疗效非劣于阿加糖酶β,并且耐受性良好,大多数不良事件为轻中度。
FDA在法布雷病治疗药物的审批中考虑患者意见,在疾病治疗效果与药物安全评估等方面都充分考虑患者的意见与诉求,推动罕见病治疗药物的上市。患者以疾病亲历者的角度提供更加精确的疾病信息和用药信息,为改进药物的研发方向和治疗方案提供了宝贵的经验与开阔的思路。同时对于审评机构而言,患者的意见也能帮助决策者进行判断,提高审批效率。


















