近期,针对天使综合征(Angelman syndrome)和1型强直性肌营养不良(DM1)这两种罕见疾病的药物研究取得了显著进展。两款药物均属于小核酸疗法,它们通过调控基因的表达来治疗疾病。

天使综合征
GTX-102是一款鞘内给药的在研反义寡核苷酸新药,旨在靶向和抑制UBE3A反义(UBE3A-AS)转录本的表达,同时激活神经元细胞中父系UBE3A的等位基因表达,产生患者体内所缺失的关键蛋白产物。
天使综合征,是由UBE3A基因缺失或突变导致的罕见性神经遗传疾病,患者会出现发育迟缓,平衡障碍、运动障碍以及癫痫发作等情况,大多不会说话。天使综合征经常被误诊为自闭症或脑瘫。目前尚未批准用于该疾病的疗法上市。
4月15日,Ultragenyx Pharmaceutical宣布了其用于治疗天使综合征(Angelman syndrome)的在研疗法GTX-102在1/2期临床试验中取得积极数据:接受GTX-102治疗的扩展队列A和B的患者,在第170天显示出具有临床意义的多领域功能改善。部分剂量递增队列患者在第758天显示出长期和持续的临床益处,远超过自然历史数据。GTX-102是一款靶向UBE3A的反义寡核苷酸疗法,通过促进神经元细胞中父系UBE3A等位基因表达,补充患者体内缺少的蛋白,达到治疗的效果。这些患者在GTX-102治疗后还表现出行为、睡眠、多动、运动和接受性沟通的显著改善,一些孩子已经能够开始自己吃饭、游泳并与家人交流。
GTX-102已经获得了美国食品药品监督管理局(FDA)的孤儿药指定、罕见儿科疾病指定和快速通道认定。
1型强直性肌营养不良
1型强直性肌营养不良(DM1)是一种进行性致命性神经肌肉疾病,患者可能会遭受一系列症状,包括肌强直和肌无力、呼吸和心脏问题、严重的胃肠道并发症,以及认知和行为障碍——导致生活质量显著下降和巨大的照顾者负担。疾病的严重程度、表现和发病年龄具有高度的变异性。
DM1 由强直性肌营养不良蛋白激酶(DMPK)基因中 CUG 三联体重复序列数量的增加引起。在健康个体中,重复序列的数量大约是35,但在 DM1 患者中,可能会有数千个。当 CUG 重复序列过多时,会形成大的发夹环,将 DMPK mRNA 困在细胞核中,有效地隔离肌肉盲样蛋白(MBNL)并降低其活性。具体来说,突变的 DMPK mRNA 结合到 MBNL,这是一种关键的 CUG 结合蛋白,形成核聚焦点,阻止 MBNL 执行其正常功能,即处理许多其他基因的 mRNA。结果,编码关键蛋白的多种 mRNA 被错误处理,导致非典型蛋白的产生,这些蛋白最终是 DM1 的病因。
Delpacibart etedesiran(AOC 1001)是一款anti-TfR1抗体偶联寡核苷酸,含有可以结合TfR1的单抗以及偶联靶向DMPK mRNA的siRNA(siDMPK.19)。TfR1抗体能够有效将AOC 1001递送到骨骼肌和心肌细胞,siRNA成分siDMPK.19则能够降低DMPK mRNA水平,进而减少细胞核中的CUG,释放MBNL,恢复正常的mRNA加工。
AOC1001治疗DM1的I/II期MARINA试验(NCT05027269)和MARINA-OLE试验(NCT05479981)最新数据显示,与END-DM1自然疾病历史数据相比,接受delpacibart etedesiran治疗的患者在多个功能指标的疾病进展出现逆转,患者的肌强直、肌肉力量和活动能力都显著改善,且安全性和耐受性良好,超过200次输注总计46.2患者年的暴露量。美国食品药品监督管理局(FDA)和欧洲药品管理局(EMA)已授予 del-desiran 孤儿药认定,FDA 还授予了 del-desiran 快速通道认定和突破性疗法认定。
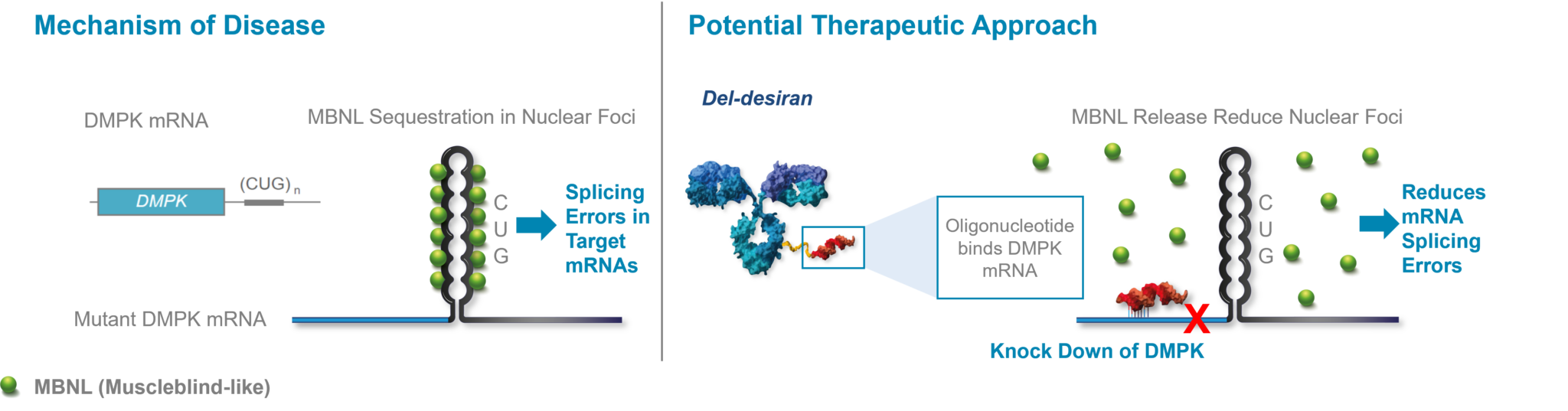
图:Avidity Biosciences官网
三期 HARBOR 试验的目的是进一步评估 AOC 1001的疗效,使用的终点包括视频手开合时间(VHOT)、肌肉力量和日常活动能力。该药物已显示出逆转疾病进展和改善患者生活质量的潜力因而获得了美国食品和药物管理局(FDA)的突破性治疗指定。


















