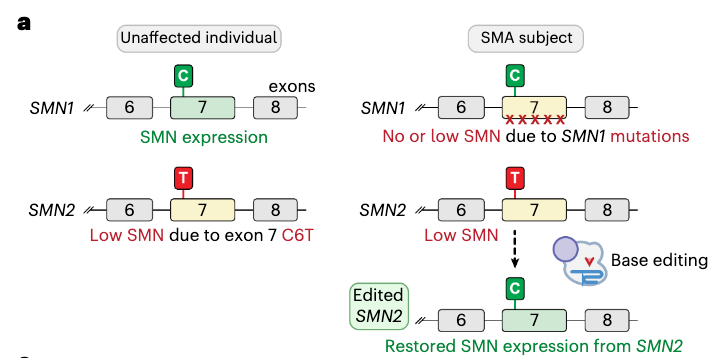
脊髓性肌萎缩症(SMA)是一种毁灭性的神经肌肉疾病,是全球婴儿死亡的重要原因。SMA的主要特征是运动神经元死亡、肌肉失神经支配和肌肉无力。大多数SMA病例是由运动神经元存活基因1(SMN1)的功能缺失突变引起的。SMN1中最常见的突变是外显子7的缺失,这导致SMN蛋白质功能丧失。
SMA严重程度的一个重要影响因素是旁系同源基因SMN2的拷贝数。SMN2的序列与SMN1不同之处在于外显子7中有一个同义的C·G-to-T·A转换。SMN2的外显子7的第6位核苷酸的C-to-T转换(简称C6T)导致大多数SMN2的mRNA中由于选择性剪接而跳过外显子7。虽然SMN2仍然产生约10%的功能性SMN蛋白质,但这不足以挽救绝大多数SMA患者。
Risdiplam(靶向SMN2的小分子药物)、Nusinersen(靶向SMN2的ASO药物)和Onasemnogene Abeparvovec(AAV基因疗法)是三种获得FDA批准上市的SMA治疗药物,这些药物已经显示出显著的患者益处,对患有SMA的新生儿的生活产生了重大影响。尽管如此,这三种疗法都存在一些问题,包括需要严格的剂量方案、存在副作用,以及长期疗效不佳。
2023年12月6日,美国哈佛医学院和麻省总医院的 Benjamin Kleinstiver团队在 Nature Biomedical Engineering 上发表了题为:Optimization of base editors for the functional correction of SMN2 as a treatment for spinal muscular atrophy 的研究论文。
该研究使用了基于SpCas9变体SpRY的腺嘌呤碱基编辑器(ABE8e-SpRY),逆转了脊髓性肌萎缩症(SMA)细胞和小鼠模型的SMN2基因的外显子7的C6T(即A-to-G编辑),从而恢复SMN蛋白水平,提高了小鼠模型的体重、运动能力和存活率。
这些结果表明,使用碱基编辑或其他基于CRISPR的基因编辑技术来靶向SMN2基因特定位点并恢复SMN蛋白水平,可能有助于SMA患者的治疗。

SMA病例的主要原因是SMN1基因的功能缺失突变,但SMN1并不是唯一参与者,SMN2基因的拷贝数是SMA严重程度的重要影响因素。SMN2基因的外显子7的第6位核苷酸的C-to-T转换(简称C6T)导致大多数SMN2的mRNA中由于选择性剪接而跳过外显子7,产生了无功能的SMN蛋白。因此,研究团队尝试使用碱基编辑技术直接编辑SMN2基因,来恢复SMN蛋白。
早在2020年,Benjamin Kleinstiver 团队就在 Science 期刊发表论文,开发了一个SpCas9的变体——SpRY,它不再需要依赖特定的PAM序列,因此能够识别并编辑基因组上几乎任何位点。
研究团队开发了基于SpRY的腺嘌呤碱基编辑器(ABE8e-SpRY),逆转SMN2基因的外显子7的C6T(即A-to-G编辑)。他们将这一编辑策略应用于从SMA患者身上提取的成纤维细胞,显著提高了SMN2的mRNA中外显子7的保留,从而提高了SMN蛋白表达水平,而且没有发现任何意外脱靶编辑。
然后,他们进一步使用双载体腺相关病毒(AAV),将gRNA和AB递送到SMA小鼠模型体内。在主要感兴趣的组织中,检测到平均约6%(大脑)和约4%(脊髓)的A-to-G编辑,这能够使SMN的转录水平显著增加2-4倍。该研究还显示,更长的随访时间会产生更高的碱基编辑水平。此外,与仅通过脑室内注射相比,额外的静脉注射能够增加对肝脏和心脏组织中碱基编辑,而且,随着时间的推移,这些治疗的小鼠模型的体重、运动功能以及生存了都有了明显改善,这说明了外周SMN蛋白的恢复对于SMA小鼠的表型恢复至关重要,可以增强其表型恢复。
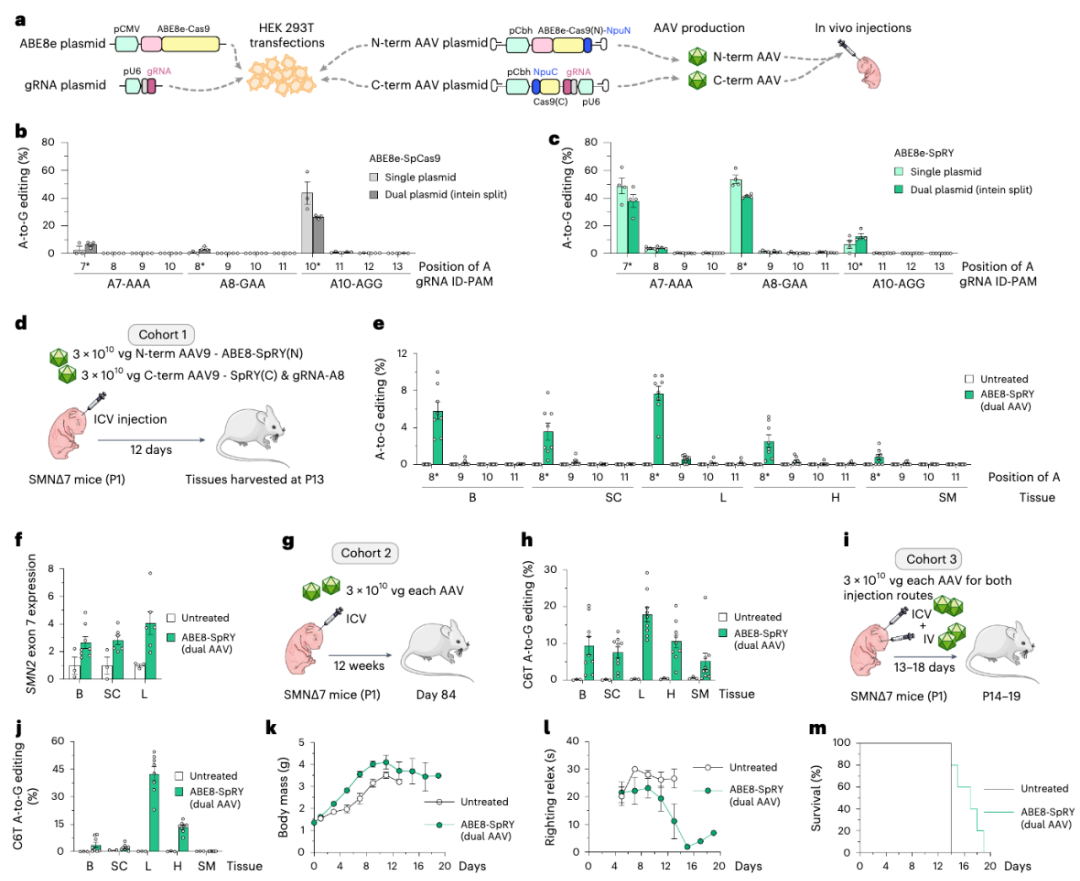
所有这些实验结果表明,碱基编辑介导的对SMN2基因的外显子7的C6T的精准编辑,能够显著增加功能性SMN蛋白水平,从而为治疗SMA提供了一种新的治疗策略。从更广泛的意义上说,这项工作证明了可以使用SpRY等适应性强的CRISPR核酸酶对基因进行精确修饰,这为使用这种方法治疗其他疾病打开了新大门。
论文链接:
https://www.nature.com/articles/s41551-023-01132-z


















