
中国Prader-Willi综合征诊治专家共识(2015)
Prader-Willi综合征(PWS)又称肌张力低下-智能障碍-性腺发育滞后-肥胖综合征、普拉德-威利综合征,由Prader等于1956年首次报道,是最早被证实涉及基因组印记的遗传性疾病。国外不同人群的发病率约为1/10000~1/30000,我国缺乏流行病学资料。PWS是症状性病态肥胖的重要病因之一,早期诊断和合理干预对改善患儿的生活质量、预防严重并发症和延长寿命至关重要。
一、临床表现
PWS的临床表现复杂多样,自胎儿期起已有异常表现、并呈现随年龄而异的时序化临床症候群,涵盖了生命过程中生长、发育、代谢等各方面。我国对儿童PWS已有临床特征研究等报道,但主要为家系或个例报道,其中相对大样本的研究提示中国PWS患者与国际上普遍描述的以西方人群为主体的临床表现不尽相同(表1)。中国的研究数据显示,PWS患儿在胎儿期活动减少,新生儿期均存在肌张力低下,婴儿期喂养困难,但只有12.9%的患者身材矮小,54.5%具有典型面容,35.5%表现出自我皮肤损伤。婴儿期营养不良是中国PWS患儿突出的表现。具有广泛代表性的中国儿童PWS临床特征谱尚待更大样本资料总结。

二、遗传机制
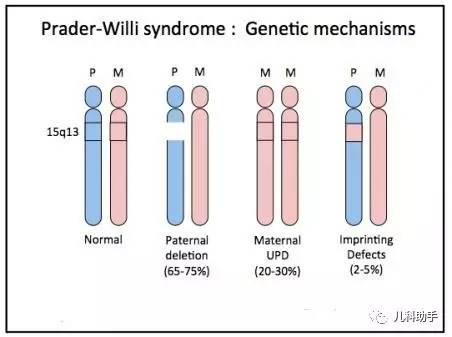
PWS为父源染色体15q11.2-q13区域印记基因的功能缺陷所致。15q11.2-q13区域长约6Mb,从染色体长臂远端至着丝粒方向可依次分为远端非印记区域、Angelman综合征印记区、PWS印记区及近着丝粒处断裂点BPl和BP2间的非印记区域4个亚区。印记中心(imprinting center)位于PWS印记区内SNURF-SNRPN基因启动子区域,掌控印记区内父源印记与母源印记之间的转换。PWS主要遗传类型包括:
1.父源染色体15q11.2-q13片段缺失(西方PWS患者占65%~75%),包括缺失I型T1D(BPl~BP3间)、缺失Ⅱ型T2D(BP2~BP3间);中国和亚洲人群该型的比例稍高于80%,要高于西方人群。
2.母源同源二倍体(UPD)导致15q11.2-q13区域的父源等位基因缺失(占20%~30%)。
3.印记中心微缺失及突变(占1%~3%)。
极少数PWS患儿(<1%)由于15号染色体发生平衡易位,尽管保留了SNURF-SNRPN基因的启动子和编码序列及其转录活性,但患儿仍呈PWS的典型表现。已有报道指出父源表达的snoRNA基因簇SNORD 116的缺失可能与PWS的表型关系密切。
三、诊断
1.临床评分诊断
中国儿童的PWS临床特征与国外不尽相同。制定PWS临床诊断评分标准时需考虑种族差异。考虑到我国此领域相关研究尚少,现阶段PWS临床评分标准,推荐仍以参考国际标准为宜。目前国际上通行的PWS临床评分标准主要根据Holm等
于1993年提出、2012年Cassidy等修正后的标准:包括6条主要标准、11条次要标准和8条支持证据。年龄<3岁总评分5分以上,主要诊断标准达4分即可诊断;年龄≥3岁总评分8分以上,主要诊断标准达5分即可诊断(表2)。

2.分子遗传诊断
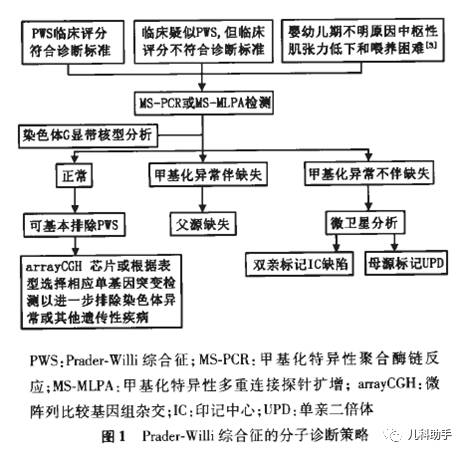
PWS临床评分诊断标准受年龄、病程、种族等多因素影响,易致漏诊或延误诊断,确诊需依据分子遗传诊断。诊断方法包括染色体核型分析技术、荧光原位杂交(FISH)、微卫星连锁分析(short tandem repeat,STR)和甲基化分析等。甲基化特异性聚合酶链反应(MS-PCR)应用早而广泛,检测符合率≥99%,但无配套试剂、操作较为繁琐,且无法区分各种缺陷类型;但其价格低廉,对于该实验条件较成熟的单位可作为筛查手段。甲基化特异性多重连接探针扩增(MS-MLPA)通过设计好的多组特异性探针可同时检测染色体多个位点的基因缺失、重复突变,结果符合率≥99%,但无法区分UPD和印记中心甲基化异常,需结合STR分析明确诊断并分型。MS-MLPA检测费用相对较贵,但有标准化配套试剂。综合国际已有经验和国情,建议根据所在实验室已有条件和经验选择相应的分子诊断方法(图1)。
四、鉴别诊断
不同年龄段的PWS表现不一,需要按照就诊相应年龄鉴别诊断。
1.婴儿期的肌张力低下需要与以下疾病进行鉴别:(1)新生儿败血症、中枢神经系统继发性异常如缺血缺氧性脑病;(2)各类神经肌肉疾病,如先天性强直性肌营养不良1型、脊肌萎缩症、先天性肌营养不良、糖原累积症2型等;(3)其他遗传综合征如Angleman综合征、脆性x染色体综合征等。
2.儿童期出现肥胖和智力异常的鉴别诊断:(1)心理性疾病等所致继发性肥胖;(2)伴有以上类似症状组分的遗传综合征如Rett综合征、Albright遗传性骨病、Cohen综合征、Bardet-Biedl综合征、Alstrom综合征、Urban-Roger综合征、Camera综合征、Vasquez综合征等;(3)染色体缺失或重复类疾病如1p36、2q37.3、6q16.2、10q26、3p25.3-26.2、xq27.2-等。
对于经MS-MLPA等甲基化分析未发现阳性结果的患儿,需结合染色体G显带核型分析及arrayCGH等分析结果,明确是否存在其他原因造成的PW样表型。
五、治疗
PWS的治疗应采用包括内分泌遗传代谢、康复理疗、心理、营养、新生儿、眼科、骨科、外科等在内的多学科参与的综合管理模式,根据不同年龄段患儿的表型特征,针对不同的内分泌代谢紊乱及相关问题进行有效干预。
1.饮食行为与营养管理
早期的饮食治疗和长期的营养监测可以改善预后。对于肌张力低下伴进食困难的婴幼儿期患儿,应尽力保证足够的热量摄人。对于吸吮无力者,可给予鼻饲管或特殊奶嘴喂养。对于年长儿,需严格管理食物,包括严格控制饮食规律,甚至将食物储存处上锁。制定三餐计划,在下一餐时间未到之前,不允许给孩子计划外的食物。尽早的饮食治疗和坚持长期的营养监测能改善预后。
对饮食行为,至今尚无一种药物可以帮助控制食欲。曾有研究应用奥曲肽(生长抑素类似物)试图降低胃饥饿素水平,结果未能改变饮食行为。胃减容手术能否用于PWS尚存争议,有报道该手术后既不能改变患儿的饱腹感,也不能改善过度摄食行为,而且手术的并发症发生率较高。根据我国目前的国情,不推荐该手术用于常规治疗。仅限于个别临床综合技术能力强的中心,在常规保守干预疗法失效的情况下,为挽救患儿极重度肥胖可能产生的致死性危险,谨慎开展探索性手术治疗。
2.性腺发育不良及青春期发育问题的处理 PWS患儿同时存在下丘脑功能低下所致低促性腺激素性性腺功能低下和原发性性腺缺陷。多数出生时即表现有性腺功能减退,但部分患儿可能迟至青春发育年龄才被发现。男性隐睾发生率近100%,小睾丸76%,阴囊发育不全69%;女性阴唇及/或阴蒂发育不全76%,56%发生原发性闭经,44%有自发性月经初潮(大多于15岁后才出现);14%有阴毛早现,3.6%发生性早熟。
(1)隐睾和外生殖器发育不良的处理和人绒毛膜促性腺激素(hCG)的应用 男性PWS性腺功能减退患儿在生后早期(<6个月)经睾酮或hCG治疗可以改善阴茎大小,促进阴囊发育,并有可能协助睾丸下降到阴囊。2014年美国泌尿外科学会共识倾向推荐此类患者采用手术疗法。如果采用hCG治疗,总量不宜超过15000 IU 。由于PWS患者的手术风险高于普通儿童,为避免手术本身以及全身麻醉呼吸并发症的风险,对于远端型隐睾,推荐可先试用hCG治疗。12月龄内患儿hCG每次250IU,1岁以上患儿hCG每次500IU,每周肌注2次,共6周,疗效不佳时仍应尽快考虑手术治疗。合适的手术时机为2岁以内,近端型隐睾以手术治疗为宜。
(2)青春期性激素替代治疗 PWS患者常需要性激素治疗以诱导、促进或维持青春发育。性激素替代治疗还对骨骼正常的发育、肌肉量的增加有积极意义,并具改善患者性生理正常化的作用。但也存在较大争议,男性患儿雄激素替代可能产生行为问题,女性患儿雌激素替代治疗可能产生月经相关的卫生问题。因此建议PWS患儿的性激素替代治疗需要与患者监护人充分讨论利弊,确定监护人意见后方可实施。
(3)性早熟的处理:约有15%~30%的PWS患儿可发生肾上腺皮质功能早现,约4%的患儿可能出现真性性早熟。但由于此类患者的性发育往往为非持续性(可自发停滞),故一般不建议采用GnRHa治疗。
3.生长激素(GH)治疗
(1)PWS开始应用基因重组人生长激素(rhGH)治疗的年龄:40%~100%PWS患儿因GH缺乏导致身材矮小。2000年美国FDA批准rhGH用于治疗PWS儿童矮小,而欧洲批准rhGH治疗PWS主要是用于改善瘦体重,而不论是否合并矮小。为此,按照美国标准PWS患儿需要达到矮小标准方可治疗,故初治年龄会偏大,而按照欧洲标准则需要早期治疗。但对于确切的起治年龄,至今为止国际上尚未有统一的指南,一般认为初治时间为婴幼儿早期、肥胖发生前(通常为2岁前)。研究发现,早期(生后3~6月龄)开始rhGH治疗还可以改善患儿精神运动发育。建议在不存在明显GH使用禁忌证的情况下,宜早于2岁开始rhGH治疗,以助肌肉组织发育、改善肌力,改善摄食能力并尽早纠正代谢紊乱情况。
(2)rhGH治疗的推荐剂量:考虑到PWS患儿在儿童期开始即可能出现超重、肥胖,因此推荐采用体表面积计算rhGH用量。起始剂量为0.5mg/(m2·d),并根据IGF-1水平(在同年龄同性别参考值的+1~+2标准差范围内)调节剂量,建议每3~6个月调整1次,逐渐增加至1.0mg/(m2·d),每日总剂量不超过2.7mg。
(3)rhGH治疗的持续时间和停药指征:rhGH治疗可一直持续至成年期,即使骨骺完全融合仍有改善体脂成分、脂代谢和认知功能的作用。推荐成年期的rhGH用量为0.1~0.2 mg/d,并使IGF-1水平维持在成年期同性别参考值的0~+2标准差范围内水平,以降低不良事件发生的概率。当存在感染和呼吸道梗阻症状时,建议暂停rhGH治疗。
(4)rhGH治疗的禁忌证及相关问题:
①禁忌证:严重肥胖、有未控制的糖尿病、未控制的严重阻塞性睡眠呼吸暂停(obstructive sleep apnea,OSA)、活动性肿瘤和活动性精神病禁用rhGH。
②需注意的相关问题:
心功能:rhGH治疗会影响心肌数量及功能,建议在治疗开始前行超声心功能检查,在长期治疗的PWS患儿中,视情复查。
胰岛素抵抗与糖尿病:rhGH治疗的PWS患儿胰岛素水平显著升高,因此在rhGH治疗的患儿中应监测糖脂代谢相关指标。
脊柱侧凸:PWS患儿的脊柱侧凸发生率较高(10岁以前30%,10岁以后80%)。尽管已有的研究未发现rhGH治疗组与对照组在脊柱侧凸、进行性侧凸的发生率上有明显差异,脊柱侧凸也并非rhGH治疗的禁忌证,但考虑到潜在的风险,推荐在rhGH治疗之前、治疗后每6~12个月进行骨科脊柱全长x线正侧位摄片检查,对比治疗前后脊柱变化情况,确定是否需要矫形治疗。
OSA:PWS儿童青少年OSA自然发生率为44%~100%,rhGH治疗可能增大舌体和腺体的体积,减小本来就狭小的气道,可能加重OSA,导致患者患上呼吸道感染时易发生猝死。对于轻中度患者扁桃体切除术后即可消失或缓解,对于PWS患者合并重度OSA,扁桃体切除术效果欠佳,因此学组建议临床实践中应注意rhGH治疗的风险和收益之间的平衡,密切、规律监测OSA症状。在出现中重度OSA情况下应暂停GH治疗,首先处理OSA再决定是否继续使用rhGH治疗。
4.其他内分泌问题的处理
约20%~30%的PWS婴儿合并甲状腺机能减退,建议左旋甲状腺素钠剂量为5~6ug/(kg·d)[<1岁,剂量为8 ug/(kg·d)],并根据游离甲状腺素和促甲状腺激素(TSH)水平调整药物剂量。
PWS患儿可发生下丘脑-垂体-肾上腺轴功能紊乱(中枢性肾上腺皮质功能低下,CAI),建议所有PWS婴幼儿在发生中重度应激事件中,都应该考虑氢化可的松替代治疗,剂量为30~70 mg/(m2·d),分3次服用。
六、遗传咨询
PWS的再发风险与其分子遗传机制有关,绝大多数PWS家庭的再发风险低于1%,但部分情况下可高达50% (表3)。PWS患儿鲜有生育的报道,其子代患PWS的概率与先证者的遗传机制及性别有关。理论上女性缺失型患者的子代有50%发生Angelman综合征的风险,而男性缺失型患者的子代有50%发生PWS的风险。由于胎盘绒毛等组织的低甲基化状态,因此不推荐将其用于产前诊断;如确实存在产前诊断的需要,可以在孕16~20周通过羊水脱落细胞的DNA甲基化分析行产前诊断。

七、随访
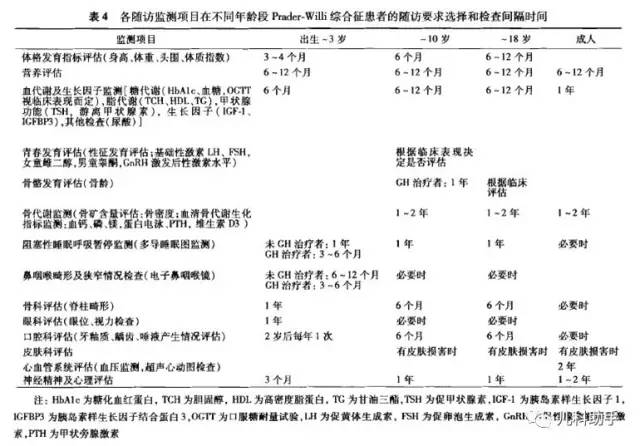
不同年龄段PWS患儿的随访指标包括体格发育、营养状况、青春发育、神经精神状况等的评估,也包括血生化指标、骨龄、骨密度、脊柱x线片等的监测,应定期进行随访观察(表4)。
资料来源:
中华儿科杂志,2015年6月第53卷第6期 419-24
中华医学会儿科学分会内分泌遗传代谢学组
《中华儿科杂志》编辑委员会
中国Prader-Willi综合征诊治专家共识(2015)


















