近日,上海交通大学医学院附属新华医院/上海市儿科医学研究所/上海市儿童罕见病诊治中心张惠文研究员团队在JAMA Network Open杂志发表了一篇题为 “Newborn Screening for 6 Lysosomal Storage Disorders in China”的研究成果,报道了上海市6种溶酶体贮积症新生儿筛查结果。该项研究成果为中国溶酶体贮积症发病率及疾病分型提供了重要的数据支撑,这是目前较大规模的关于溶酶体贮积症新生儿筛查的研究成果。

一、研究背景及目的
溶酶体贮积症(LSDs)是一类由基因突变导致溶酶体水解酶或转运蛋白异常导致其底物在细胞内外堆积所引起的疾病。LSDs包含近60种类型,大部分为常染色体隐性遗传,部分为X染色体连锁遗传。据2022年美国医学遗传学与基因组学学会的报告,LSDs的总发病率为1/4 000~1/9 000。LSDs的治疗方法主要包括骨髓移植、酶替代疗法、底物减少疗法、分子伴侣疗法和基因疗法等。早期诊断及治疗,对降低疾病相关的致死致残率至关重要。意大利、美国、日本等,已纳入部分LSDs为新生儿筛查项目。本研究利用串联质谱法(MS/MS)筛查6种LSDs,包括戈谢病、尼曼匹克病A/B型、克拉贝病、黏多糖贮积症I 型、法布雷病和庞贝病,较准确地评估了上海市6种LSDs发病率情况并判断了患儿的临床分型。
二、研究列队
此队列研究纳入50 108例新生儿,通过MS/MS法同时检测新生儿干血滤纸片样本中6种LSDs酶活性,包括酸性β-葡萄糖脑苷脂酶(ABG;戈谢病)、酸性鞘磷脂酶(ASM;尼曼匹克A/B型)、β-半乳糖脑苷脂酶(GALC;克拉伯病)、α-L-艾杜糖醛酸酶(IDUA;粘多糖贮积症I 型)、 α-半乳糖苷酶A(GLA;法布雷病)、酸性α-葡萄糖苷酶(GAA;庞贝病)。以每批次检测结果6种酶活性水平中位数值的20%作为该次酶活性的切值。对筛查阳性的新生儿进行基因分析及生化标志物检测,并由遗传代谢科医生对筛查阳性新生儿进行诊断并确认临床表型。
三、LSDs新生儿筛查
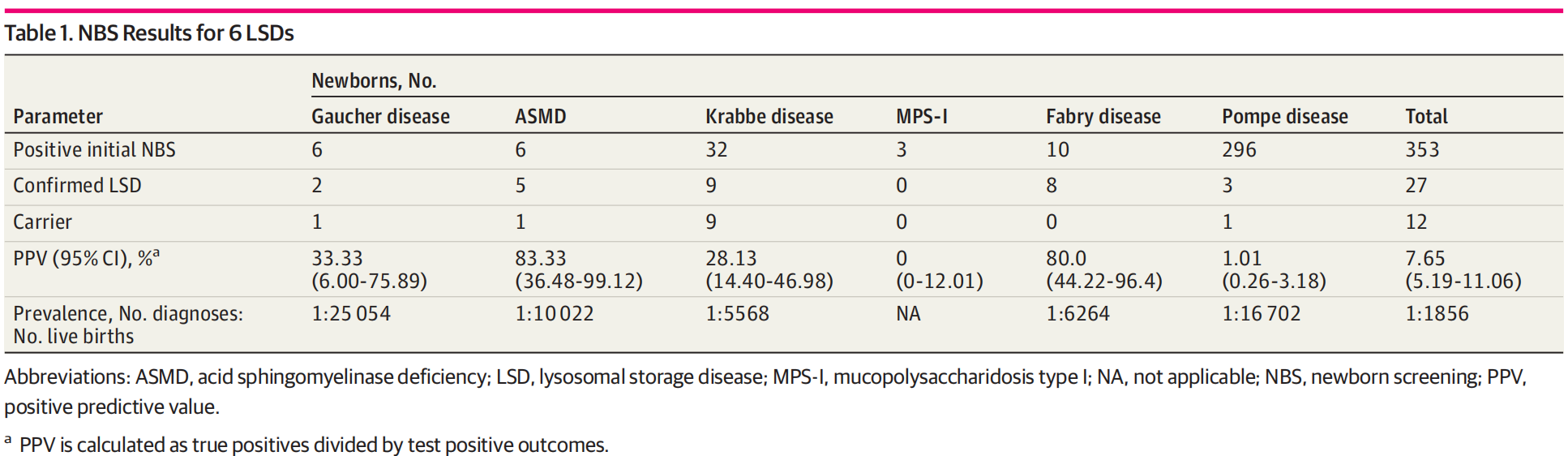
353例酶活性水平低于切值,初筛阳性率为0.70%(353/50 108)。经基因检测和生化标志物检测及临床分析后,共确诊27例LSDs,包含2 例戈谢病、5 例尼曼匹克A/B型、9例克拉伯病、8例法布雷病和3例庞贝病。上海市 LSDs总发病率为 1/1 856,戈谢病、尼曼匹克A/B型、克拉贝病、法布里病和庞贝病发病率分别为 1/25 054、1/10 022、1/5 568、1/6 264 和 1/16 702。
四、分子遗传特征及生物标志物分析
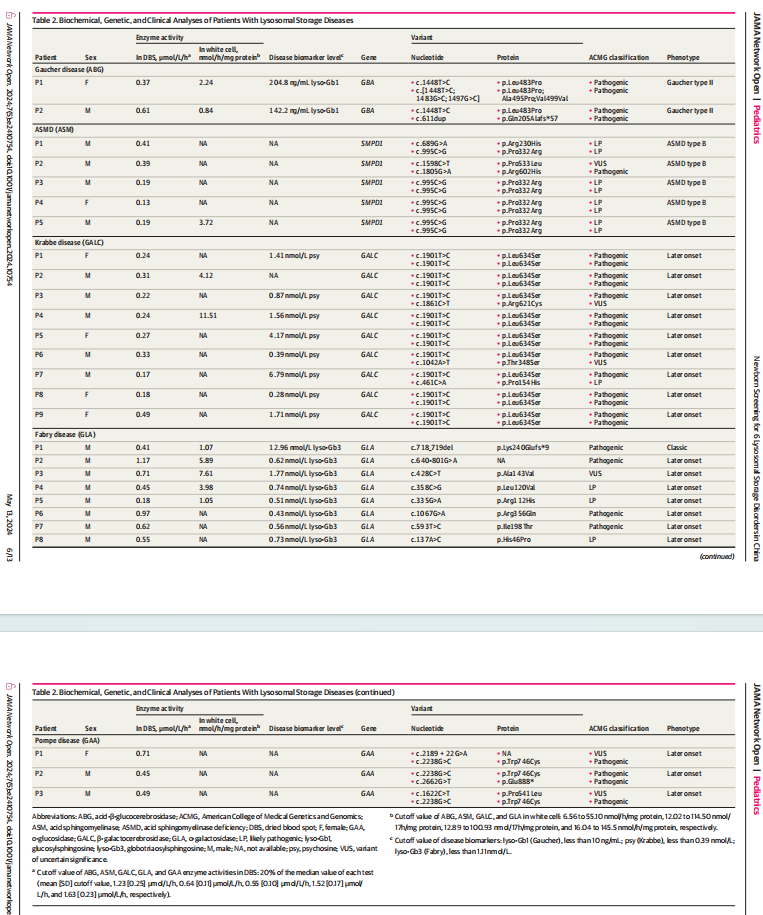
戈谢病初筛阳性新生儿6例,确诊2例,均检出GBA基因变异。最常见的变异体为c.1448T>C(p.Leu483Pro),是一种与神经系统改变有关的致病变异。2例患儿生物血浆标志物Lyso-Gb1浓度显著升高,为173.5±44.29 ng/mL(切值<10.0 ng/mL)。基于上述结果,这2例患儿为神经系统受累最严重的戈谢病Ⅱ型。
尼曼匹克A/B型初筛阳性新生儿6例,确诊5例,均检出SMPD1基因变异。最常见的变异体为c.995C>G (p.Pro332Arg),5例患儿中有4例检出此变异。c.995C>G (p.Pro332Arg)纯合型变异与成人型尼曼匹克B型相关。这5例患儿分型为尼曼匹克B型。
克拉伯病初筛阳性新生儿32例,确诊9例,是6种LSDs中最常见的类型,占比33.3%(9/27)。患儿均检出GALC 基因变异,6例为c.1901T>C(p.Leu634Ser)纯合型变异,3例为c.1901T>C (p.Leu634Ser)和其他错义突变的复合杂合型变异。最常见的变异体为c.1901T>C(p.Leu634Ser),在全部等位基因的占比为83.3%,其纯合型可能与轻型成人型表型相关。文献报道,血浆标志物Psychosine可作为本病的二级筛查,将患儿分为早发型和晚发型克拉伯病。GALC酶活性缺乏的新生儿,Psychosine浓度在2~10 nmol/L之间可归为晚发型克拉伯病,而浓度大于10 nmol/L可推测为早发型克拉伯病。本研究6例患儿出生3天时Psychosine浓度低于2 nmol/L,2例浓度在2~10 nmol/L之间。因此,这9例患儿均为晚发型克拉伯病。
黏多糖贮积症I型初筛阳性新生儿3例,2例检出IDUA基因变异,分别为c.164C>A (p.Pro55Gln)/c.1093C>G (p.Leu365Val)/c.1828+5G>A和c.355G>T (p.Asp119Tyr)/c.911del (p.Val304Glyfs*13),1例未检出IDUA基因变异。2例基因检测结果呈阳性的新生儿尿黏多糖定性分析为阴性,电泳检测结果未见硫酸皮肤素和硫酸类肝素条带。因此,这3例新生儿排除为黏多糖贮积症I型,为假阳性。
X连锁的法布雷病初筛阳性新生儿10例,确诊8例,均为男性,在所有男性新生儿中约占比1/3 000。8例患儿均检出GLA基因变异:1例检出经典型变异,c.718_719del (p.Lys240Glufs*9);6例检出晚发型变异。经典型变异的患儿血浆标志物Lyso-Gb3浓度显著升高,出生3天时浓度超出正常参考范围上限的12 倍,4月时增至24倍(12.96 和 26.83 nmol/L,切值< 1.11 nmol/L)。患儿P3出生3天时Lyso-Gb3水平(1.77 nmol/L)轻度升高,召回时增至 4.43 nmol/L。另外6例患儿出生3天时Lyso-Gb3水平在正常范围内,分别为0.62、0.74、0.51、0.43、0.56 和 0.73 nmol/L,其中2例在随访时浓度增至3.42 和 2.73 nmol/L(随访时年龄为10月和4月)。基于上述结果,仅1例患儿归为经典型,其余7例归为晚发型。
庞贝病初筛阳性新生儿296例,初筛阳性率高达0.59%。此高阳性率主要与假性缺陷等位基因(如:c.[1726G>A; 2065G>A])在亚洲人群中的携带率较高有关。既往研究显示,在日本、台湾及山东分别有5.6%、8.7和1.1%初筛阳性新生儿被诊断为庞贝病。山东确诊的3例庞贝病患儿,GAA酶活性低于每批次酶活性中位数值的6%,而本院5例临床患者酶活性水平约为中位数值的5%。因此,本研究未对GAA酶活性水平在中位数值10%~20%之间的筛查阳性新生儿进行基因检测,仅对酶活性水平低于中位数值10%的新生儿进行基因检测。基因结果显示,3例患儿均含有在中国庞贝病患者中常见的晚发型GAA致病变异,c.2238G>C (p.Trp746Cys)。此3例患儿为晚发型庞贝病。
小结
研究表明, MS/MS新生儿酶活性筛查联合基因检测和生物标志物检测及临床分析,成功确认了患儿的临床亚型。上海市常见LSDs综合发病率a高达1/1 856,晚发型占比相对较高。中国人群部分基因存在热点突变,导致对应疾病发病率相对较高,比如GALC 基因c.1901T>C(p.Leu634Ser)变异,SMPD1基因c.995C>G (p.Pro332Arg) 变异,GAA基因c.995C>G (p.Pro332Arg)变异,GBA1基因c.1448T>C (p.Leu483Pro) 变异。
原文链接:Chang S, Zhan X, Liu Y, Song H, Gong Z, Han L, Maegawa GHB, Gu X, Zhang H. Newborn Screening for 6 Lysosomal Storage Disorders in China. JAMA Netw Open. 2024 May 1;7(5):e2410754.


















