目的
报道NPC1基因新发复合杂合突变致尼曼-匹克病C型一家系的临床及遗传学特点,提高临床医师对该病的认识水平。
方法
对云南省昆明医科大学第一附属医院神经内科2020年收治的1个非近亲结婚家系中的2例患者进行详细的神经科体格检查,提取外周血DNA,应用二代测序技术对患者进行全外显子测序,结合Sanger测序对患者家系进行一代验证,应用软件对突变位点进行分析。
结果
该家系呈常染色体隐性遗传,2例患者的发病年龄不同,先证者9岁起病,主要表现为:构音不良、吞咽困难、认知障碍、共济失调、双侧锥体束损害、垂直核上凝视麻痹和脾脏肿大。先证者弟弟临床表型与患者相似,但比患者更为严重,发病年龄更早,婴幼儿期发病,伴有严重的精神运动发育迟缓。全外显子组测序结果显示,两兄弟均携带NPC1基因的两种罕见变异:c.352_353del,p.Gln119ValfsTer8和c.593A>G,p.Asn198Ser。Sanger测序验证结果提示复合杂合突变分别来源于先证者的父母。根据美国医学遗传学与基因组学学会指南,上述变异评级分别为致病和疑似致病变异。该家系患者携带的c.593A>G,p.Asn198Ser变异为未报道的新变异。先证者从症状出现开始延迟诊断7年,服用麦格司他1年后吞咽困难、共济失调及眼球垂直运动障碍等症状有明显的好转。
结论
该家系患者的临床表型符合尼曼-匹克病C型的临床表型,NPC1基因复合杂合突变(c.352_353del和c.593A>G)为该家系的遗传学病因。
尼曼-匹克病C型(Niemann-Pick disease type C,NPC)是一种以进行性神经变性为特征的常染色体隐性遗传性脂质沉积病,由Crocker和Farber[1]于1958年首次描述,由NPC1和NPC2基因突变引起,可导致胆固醇和脂质从溶酶体和晚期核内体的转运受损,导致脂质在受累细胞内蓄积。NPC具有高度的临床和遗传异质性,加之临床医师对该病的认识不足,常常导致漏诊、误诊及诊断延迟[2, 3]。NPC是为数不多的可治性遗传代谢性疾病,麦格司他是治疗该病的特异性药物,在神经系统症状出现前开始治疗的患者预后更好,因此,早期及时准确诊断该病尤为重要[4, 5, 6]。文中就1个NPC家系的临床特征及基因突变特点进行分析,旨在提高临床医师对此罕见疾病的认识,减少临床误诊、漏诊及延迟诊断。
资料和方法
一、临床资料
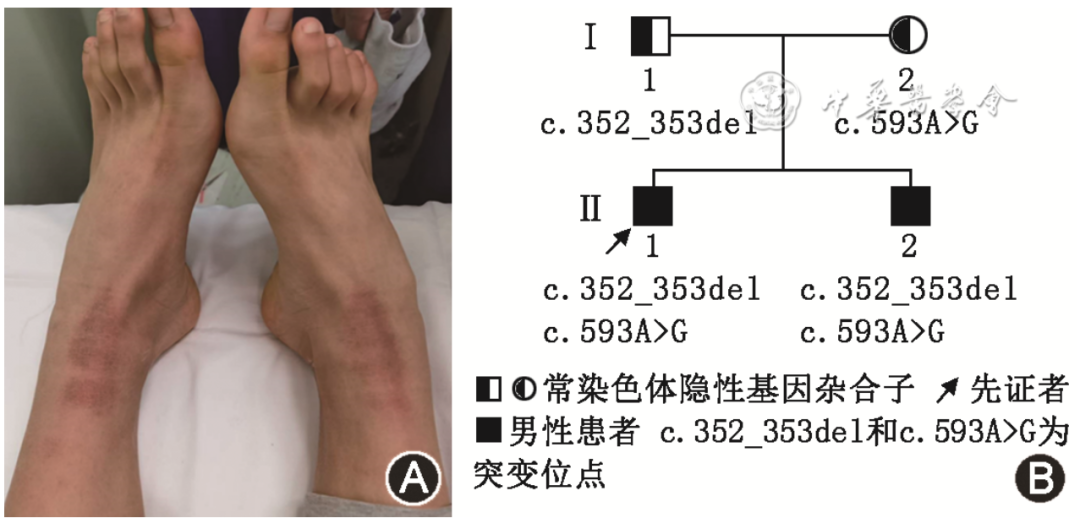
先证者男性,15岁,因“反应变慢7年,言语不清、行走不稳1年”于2020年8月就诊于昆明医科大学第一附属医院神经内科。先证者9岁时出现反应变慢、学习成绩逐步下降(从前十名逐步滑落到最后一名)。11岁起经常出现“梦游”症状,曾多次在儿科及儿童医院就诊,未能明确诊断。14岁时无明显诱因出现言语不清、说话变慢、吞咽困难、双上肢动作笨拙和行走不稳。入院前1个月,患者上述症状加重并经常跌倒。既往史:有7年前“阑尾切除”手术史,余无特殊。家族史:父母体健,否认近亲结婚,患者的祖辈身体健康,否认类似病史。先证者弟弟症状、体征与患者相似,但症状更重,自幼智力、语言及运动发育均落后于同龄儿童,现7岁,仅能讲简单的字词,能走,但不能跑,容易跌倒,曾在当地儿童医院诊断“精神发育迟滞”。先证者神经系统体检:构音不良、吟诗样语言、反应慢、记忆力及计算力轻度减退,双侧瞳孔等大等圆,直径约3 mm,直接及间接对光反应灵敏,双眼球垂直运动障碍,下视显著受限,左右视时引出水平眼球震颤;四肢肌张力减低,肌力正常,四肢腱反射亢进,双侧踝阵挛(+),双下肢查多克征(+),双侧指鼻试验、双下肢跟膝胫试验欠稳准,Romberg征睁眼及闭眼均阳性,高足弓,马蹄内翻足畸形(图1A)。辅助检查:血尿便常规、肝肾功能、血糖、血脂、心肌酶、凝血功能、抗核抗体谱、抗磷脂抗体、免疫球蛋白及补体、肝炎、梅毒及HIV等检查未见明显的异常。铜蓝蛋白:0.14 g/L,眼底裂隙灯检查未见K-F环。甲状腺功能:甲状腺微粒体抗体116.30%(参考值<10.00%),甲状腺球蛋白抗体301.40%(参考值<95.00%)。心脏超声:二维超声心动图及彩色血流检查正常。腹部超声:脾肿大,厚径5.3 cm,直径12.3 cm,实质回声均匀,其内未见异常结构。甲状腺超声:甲状腺实质回声不均声像,桥本病可能。胸CT平扫:右肺中叶实性微结节。头颈胸椎磁共振成像未见明显异常。四肢肌电图:所检神经的传导速度及F波未见异常。
图1 尼曼-匹克病C型先证者的足部畸形和家系图。A:先证者高足弓,马蹄内翻足畸形;B:NPC1基因复合杂合突变C.352_353del 和 C.593A>G导致常染色体隐性遗传的尼曼-匹克病C型家系图
Figure 1 Foot deformities of the proband with Niemann-Pick disease type C and family pedigree chart. A: The proband had a high arch, clubfoot deformity. B: Pedigree of the autosomal recessive Nieman-Pick disease type C caused by compound heterozygous mutation of NPC1 gene C.352_353del and C.593A>G
二、研究方法
1.神经心理学检查:利用简易精神状态检查量表(Mini-Mental State Examination,MMSE)、蒙特利尔认知评估量表(Montreal Cognitive Assessment,MoCA)及日常生活能力量表(Activity of Daily Living Scale,ADL)评估先证者整体认知功能及日常生活能力。
2影像学检查:对先证者进行头颅、颈胸椎MRI检查,了解颅内及脊髓的病变情况。
3.基因检查:对先证者及其弟弟和父母(图1B)进行基因检测。具体方法为:经监护人知情同意后抽取先证者及家系成员外周静脉血4 ml,提取基因组DNA并进行全外显子测序(委托北京康旭有限责任公司进行测序),对发现的可疑致病性基因变异进行Sanger测序,并在家系成员中进行验证。
本研究为家系研究,经昆明医科大学第一附属医院医学伦理委员会批准[批件号:(2022)伦审L第159号],并获得患者监护人书面知情同意。
结果
一、神经心理学评估
先证者MMSE评分16分(定向力5分+记忆力3分+注意力和计算力2分+回忆能力1分+语言能力5分),MoCA 评分9分(视空间与执行功能0分+命名1分+注意3分+语言1分+抽象0分+延迟回忆1分+定向力2分,受教育年限≤12年,加1分),ADL评分36分。先证者弟弟不能配合完成MMSE和MoCA测评。
二、影像学检查
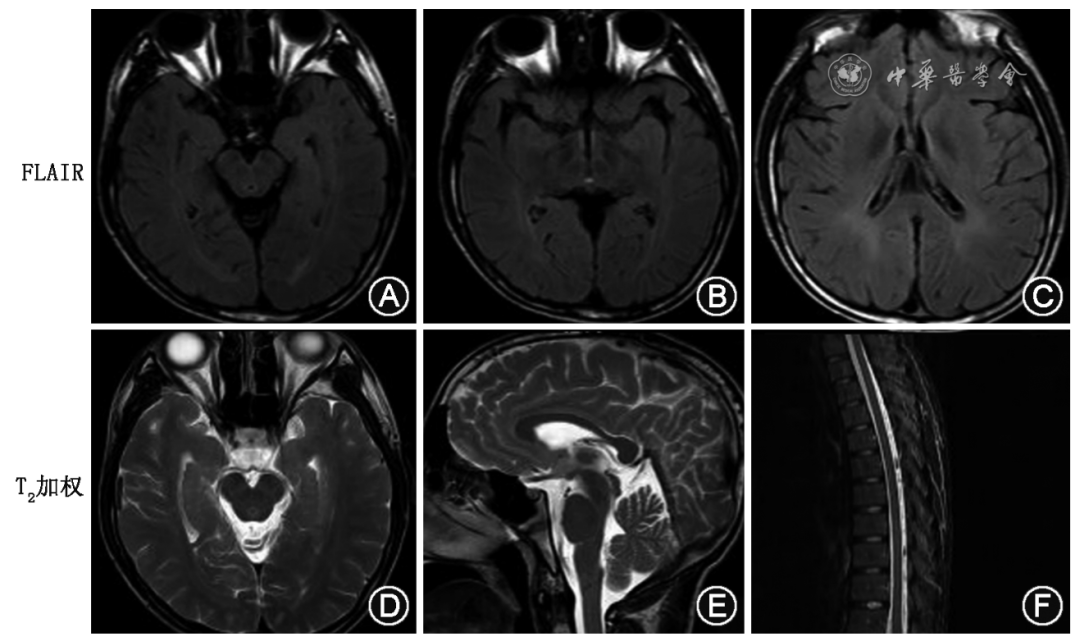
先证者头颅MRI示:在FLAIR像见中脑导水管、第三脑室周围和侧脑室旁白质信号增高(图2A~C)。T2加权像示四叠体池及脚尖窝增宽,胼胝体可疑变薄,胸段脊髓可疑变细(图2 D~F)。
图2 尼曼-匹克病C型先证者头颅磁共振成像。在液体衰减反转恢复(FLAIR)像(A~C)见中脑导水管、第三脑室周围和侧脑室旁脑白质信号增高。T2加权像示四叠体池及脚尖窝增宽,胼胝体可疑变薄(D、E),胸段脊髓可疑变细表现(F)
Figure 2 Brain magnetic resonance imaging of the proband with Niemann-Pick disease type C. Increased signal intensity of white matter around the lateral ventricle, the mesencephalic aqueduct and periphery of the third ventricle was shown on fluid attenuated inversion recovery (FLAIR) images (A-C). T2-weighted imaging showed widening of the quadrigeminal cistern and apex fossa, suspicious thinning of the corpus callosum (D,E), and suspicious thinning of the thoracic spinal cord (F)
三、基因检测及变异分析
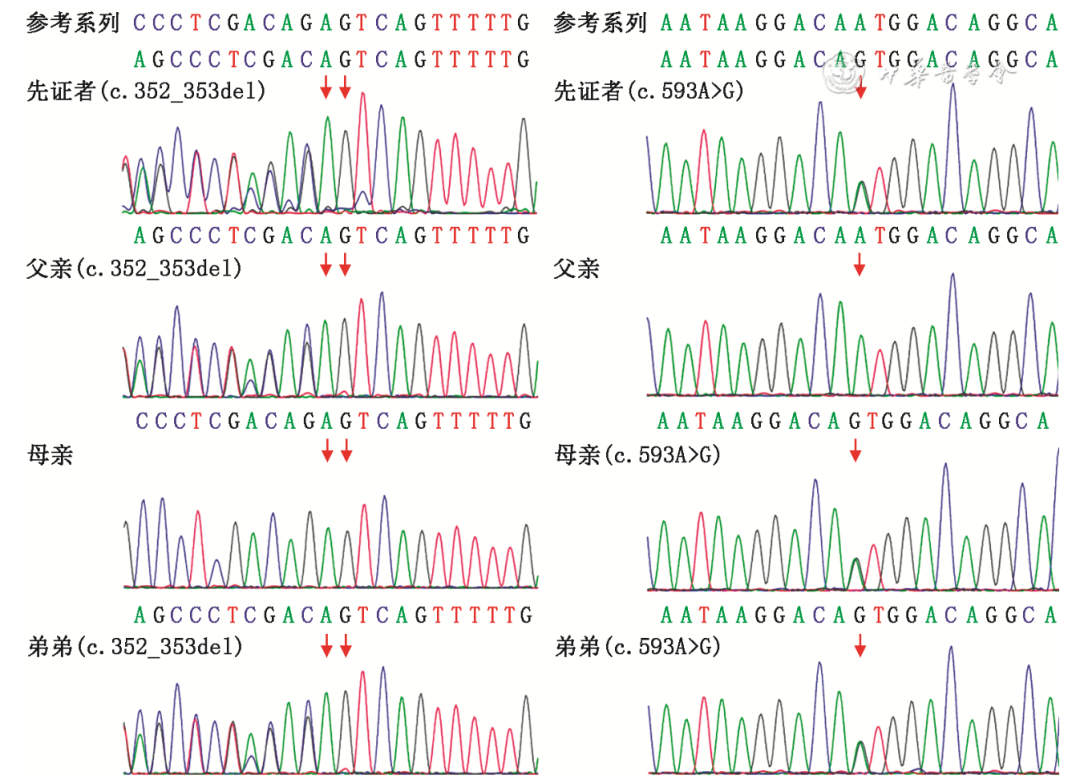
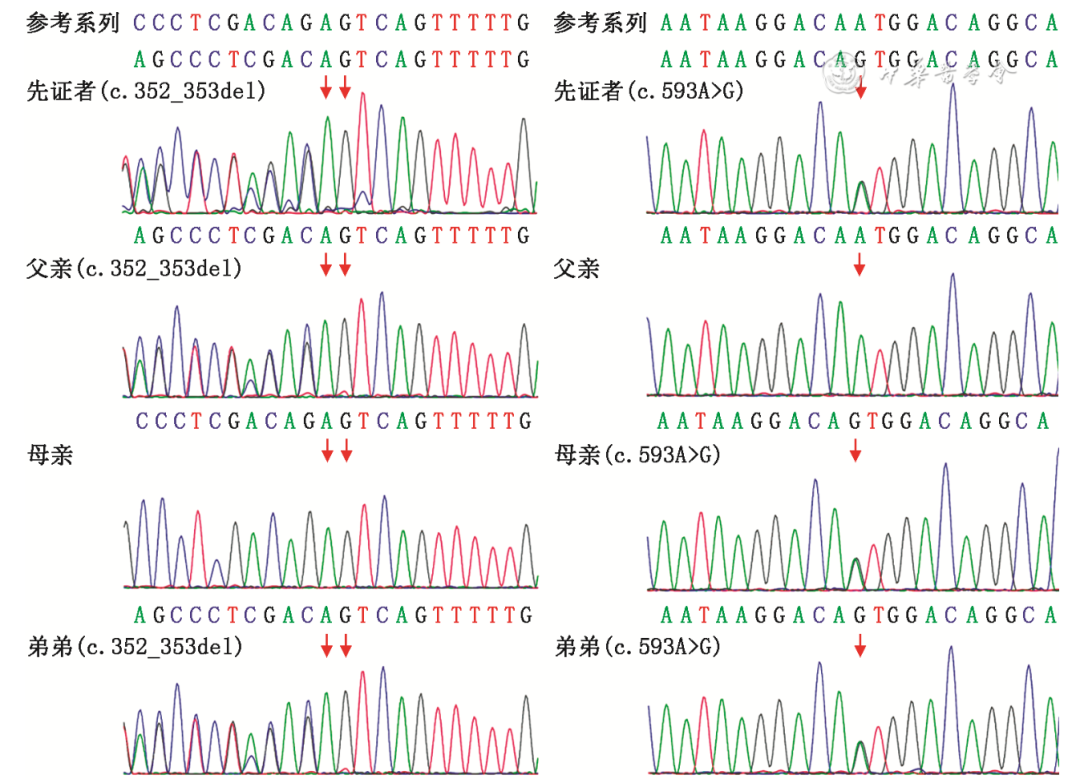
三人全外显子基因测序结果显示先证者NPC1基因存在2个变异位点:c.352_353del、c.593A>G(NM_000271),c.352_353del源自父亲,c.593A>G源自母亲,构成复合杂合突变,符合常染色体隐性遗传规律,并经Sanger测序进行了家系验证(图3)。并通过SIFT Pred、Polyphen-2 HVAR Pred、MutationTaster Pred软件进行基因有害性预测。根据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)遗传变异分类标准与指南,c.352_353del 为已报道的移码突变,该变异引起基因开放阅读框发生改变,导致蛋白功能改变,在人类外显子数据库(ExAC)、参考人群千人基因组(1000G)和人群基因组变异频率数据库(gnomAD)中均未见收录,与一个已知的致病性变异组成复合杂合变异的隐性遗传,致病变异证据:PVS1+PM2+PM3。c.593A>G为错义变异,该变异ACMG证据为可能致病性变异:PM1+PM2+PM3+BP4。经查询人类基因组突变数据库(HGMD)、PubMed、ClinVar等相关专业数据库,未见相关文献报道及收录。先证者弟弟的基因检测结果提示突变位点与先证者一致(图3)。
图3 PNC1基因复合杂合突变导致尼曼-匹克病C型患者家系的Sanger测序验证图(红色箭头提示突变位点所在位置)
Figure 3 Sanger sequencing for the patients of a family with Niemann-Pick disease type C caused by compound heterozygous mutations in the PNC1 gene (The red arrows indicate the location of the mutation site)
四、临床特点及治疗随访
该家系为常染色体隐性遗传模式。先证者为青少年型,症状体征较轻,主要临床表型:构音不良、吞咽困难、认知障碍、共济失调、双侧锥体束损害、垂直核上性凝视麻痹和脾脏肿大,高足弓和马蹄内翻足畸形。先证者弟弟为晚期婴幼儿型,除上述表型外伴有严重的精神运动发育迟滞。经基因检测证实该家系为NPC。先证者口服麦格司他200 mg,每天3次;治疗后1年随访,先证者的认知功能、言语不清、吞咽困难、行走不稳、跌倒和垂直性核上性凝视麻痹症状均有明显的缓解。患者弟弟因为症状较重,加之家庭经济原因一致未服药治疗。
讨论
NPC是慢性、危及生命的遗传代谢性疾病,在过去20年,由于致病基因和潜在代谢通路被发现,以NPC为代表的超罕见遗传代谢性疾病受到了越来越多的关注,这使得NPC靶向治疗成为可能。此类疗法可对病程产生重大影响,可提高患者生活质量并改善结局,但通常需要早期和迅速启动治疗,以尽量减少或预防不可逆的病理改变[5,7]。因此,及时准确的诊断至关重要。本研究中的2例患者,携带NPC1基因复合杂合突变(c.352_353del和c.593A>G),其中c.593A>G,p.Asn198Ser变异为未报道的新变异,丰富了NPC1基因突变谱,为遗传咨询和产前诊断提供了依据。
本家系中的2例患者,起病年龄和临床表型有很大的差异,这与既往的研究报道一致,NPC具有显著的临床和遗传异质性[2]。据报道,NPC的主要临床特征包括小脑性共济失调、垂直核上性眼肌麻痹、构音障碍、认知障碍、运动障碍、脾肿大、精神障碍和吞咽困难,较不常见的症状为癫痫、猝倒和皮质肌阵挛发作等[8, 9, 10, 11, 12, 13]。垂直性核上性凝视麻痹是NPC具有特征性的临床表现之一,临床医生应加强对此体征的识别,有助于为NPC早期诊断提供有用的线索[10,14, 15]。本研究中的2例患者都有明确垂直性眼球活动障碍,尤其是下视受限明显。先证者头颅MRI提示中脑导水管、第三脑室周围白质的异常信号,四叠体池和脚尖窝的增宽提示中脑萎缩可能,这可能是导致患者出现垂直性核上性麻痹的原因。先证者侧脑室旁脑白质的信号异常,胼胝体变薄和脊髓的变细可能与患者的认知障碍、锥体束受损、共济失调、构音障碍相关。先证者的脑白质信号的异常和胼胝体变薄与既往的报道一致[3,16],但无特异性,对诊断帮助不大。NPC除有神经系统受累外,常伴有内脏系统受累表现,如肝脾肿大和间质性肺炎等[17],骨髓、肝、脾、肺等组织病理检查发现富含脂质的巨噬细胞,也称泡沫样细胞或尼曼-匹克细胞有助于诊断。近年来,血浆壳三糖苷酶、7-酮胆固醇、胆甾醇-3β、5α、6β-三羟基胆酰甘氨酸和胆汁酸等已成为 NPC特异性生物诊断标志物,但基因检查仍被认为是NPC确诊的有效手段,并可以和其他遗传代谢性疾病相鉴别[18, 19, 20]。如先证者铜蓝蛋白降低,但眼科裂隙灯检查未看见K-F环,基因检测未发现ATP7B基因的致病突变,可排除肝豆状核变性;先证者体格检查可见锥体束征、高足弓、马蹄内翻足畸形,MRI显示胼胝体可疑变薄、胸段脊髓可疑变细表现,临床需要与伴有变薄胼胝体的遗传性痉挛性截瘫进行鉴别,但基因检测并未发现遗传性痉挛性截瘫的致病基因,可排除该病。NPC的确诊需要临床医师进行详细的病史询问、细致的体格检查,一旦临床怀疑NPC,可通过生物标志物检测、骨髓穿刺活组织检查、皮肤成纤维细胞filipin染色和基因检测进行确诊。正如本家系中的先证者一样,以麦格司他为代表的药物治疗是成功的,服用麦格司他治疗1年后,先证者的神经症状有明显的改善。但不幸的是,该药目前价格仍然较贵,虽然已经进入国家的谈判药系列,报销比例已经大幅提高,但很多家庭仍无力承担,先证者弟弟因为经济原因一直未服药治疗。除麦格司他外,环糊精、双嘧达莫、N-乙酰-L-亮氨酸和阿莫洛莫在相应的临床试验中也显示出有效性和安全性[15,21, 22]。
总之,NPC是为数不多的有药物可以有效干预的遗传代谢性疾病,提高临床医师对该病的认识水平、及时准确的诊断、合理的药物治疗是改善患者生活质量和预后的有效策略。