ChenFJMU内容团队编辑(原创,文末蔡磊《相信》书籍推荐及读后感分享)

ALS、SMA、DMD、FRDA四种神经肌肉罕见病ASO药物新盘点
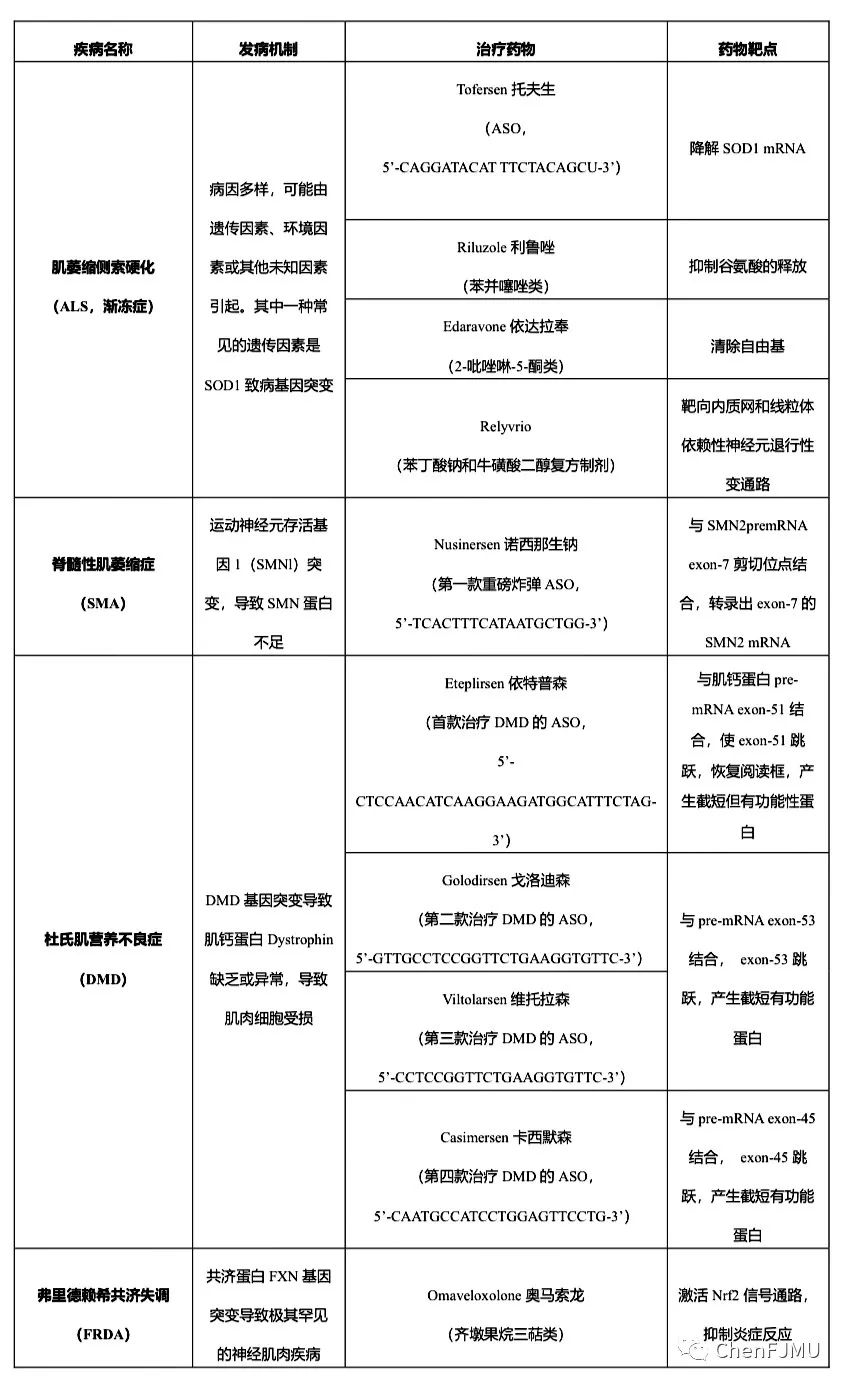
肌萎缩侧索硬化(渐冻症,ALS)、脊髓性肌萎缩症(SMA)、杜氏肌营养不良(DMD)、弗里德赖希共济失调(FRDA)四种神经肌肉罕见病,主要表现为肌肉无力,肌肉萎缩,行动费力,逐渐加重甚至瘫痪,部分伴有肌肉疼痛、痉挛等表现,甚至累及心肌、呼吸肌危及生命。FDA共批准6种反义核酸药物(ASO),其中Tofersen用于治疗ALS;Nusinersen用于治疗SMA;4个ASO药物(Eteplirsen、Golodirsen、Viltolarsen、Casimersen)用于强攻DMD。本文将着重盘点用于这四种神经肌肉罕见病的ASO药物,涵盖3种作用机制:RNase H介导的SOD-1基因沉默、改变SMN-2 pre mRNA剪切(使其含外显子7)、外显子跳跃技术(跳跃外显子51/53/45)。
01“渐冻症”新药里程碑:首款靶向SOD1反义核酸药物Tofersen上市!
02一针70万变3万!第一款重磅炸弹ASO药物Nusinersen治疗SMA。
03四款外显子跳跃技术ASO药物上市,DMD治疗按下“加速键”
04又一款“孤儿药”,弗里德赖希共济失调患者无药可治成为过去式
一、治疗渐冻症ALS药物新盘点(ChenFJMU)
Tofersen,01
Qalsody(Tofersen,托夫生)由美国Biogen和Ionis公司联合研发,FDA于2023年4月25日通过加速批准上市,用于治疗具有超氧化物歧化酶1突变的肌萎缩侧索硬化(SOD1-ALS)(俗称“渐冻症”),是首款ALS反义核酸药物,也是FDA批准第四款治疗ALS药物。
渐冻症病因多样,可能由遗传因素、环境因素或其他未知因素引起。其中一种常见的遗传因素是SOD1致病基因突变。早期五大症状为:肌无力、肌萎缩、吞咽困难、饮水咳呛、言语不清。随着疾病的发展,肌肉萎缩越发的严重,最终出现肌无力和运动功能丧失。
【结构与剂型处方】
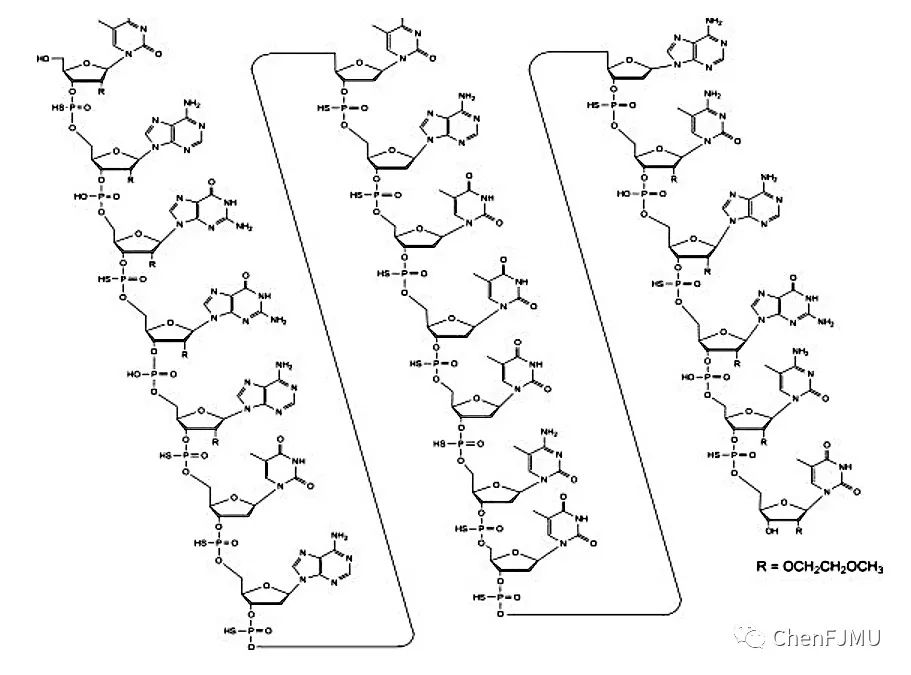
Tofersen是一种反义寡核苷酸药物(ASO),序列为:5’-CAGGATACAT TTCTACAGCU-3’(20-mer,15个硫代磷酸酯键PS,4个磷酸二酯键)。Tofersen是MOE PS gapmer嵌合反义核酸(5-10-5):5'和3'-端分别有5个2'-O-甲氧基乙基-核糖核苷酸(2'-O-MOE),中间是10个2'-脱氧核糖核苷酸组成的间隔。化学式是C230 H317 N72 O123 P19 S15;分子量是7127.86。Tofersen以注射剂的形式装在玻璃小瓶中,通过鞘内注射给药。每瓶含有单剂量为100 mg 的Tofersen,浓度为6.7 mg/mL。Tofersen的pH值约为7.2(范围为6.7-7.7)。
【作用机制】
超氧化物歧化酶1(SOD1)基因是第一个发现的ALS致病基因,SOD1基因突变会导致抗氧化酶作用减弱和线粒体功能障碍。Tofersen是一款靶向治疗成人SOD1基因突变型 ALS的反义核酸药物。Tofersen 能降解 SOD1 mRNA,减少 SOD1 蛋白质的合成,并使人体清除现有的SOD1 蛋白质,从而保护运动神经元完整性,达到治疗渐冻症的目的。Tofersen(MOE PS gapmer结构)通过核糖核酸酶RNase H对DNA-RNA杂交体的降解达到治疗过程中沉默SOD1基因的目的。Gapmer通过结合靶mRNA,使得gapmer DNA-mRNA杂交双链被RNase H降解,导致靶mRNA被降解,从而阻止SOD-1蛋白合成。Gapmer结构使得Tofersen增加对核酸酶的抗性,提高体内稳定性;还提高与靶mRNA的亲和力,减少了脱靶效应、非特异性结合和不必要的基因沉默。
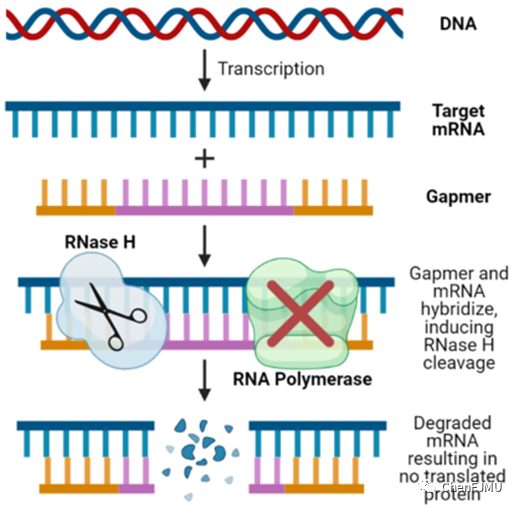
Tofersen(MOE PS gapmer结构)作用机制图
【药效学及药代动力学】
药效学:Tofersen使得脑脊液SOD1蛋白总量减少;Tofersen治疗使得患者血浆NfL含量降低(血浆NfL神经元蛋白是一种基于血液的轴索损伤和神经变性biomarker);Tofersen不会延长QTc间期。
吸收:Tofersen经鞘内给药使其进入脑脊液,并从脑脊液分布到中枢神经系统组织。脑脊液中最大浓度出现在第三次给药时。Tofersen给药后血浆中到达峰浓度所需的时间为2-6 h。
分布:鞘内使用的Tofersen分布在中枢神经系统组织内。
消除:Tofersen通过核酸外切酶(3'-和5'-)介导代谢;Tofersen不通过CYP450酶代谢,也不是其抑制剂或诱导剂。尚未确定主要的消除途径。脑脊液中半衰期为4 wk。
药物相互作用:尚未进行临床药物相互作用研究。Tofersen不是CYP酶主要的底物或抑制剂/诱导剂,也不是转运蛋白的主要底物或抑制剂。
特定人群:未进行临床研究评估Tofersen在肾脏或肝脏损伤患者的药代动力学。
免疫原性:与所有治疗性寡核苷酸药物一样,Tofersen存在潜在的免疫原性。在患者中通过检测抗药物抗体(ADA)评估Tofersen的免疫原性反应。Tofersen患者出现治疗性ADA,其存在使血浆Tofersen清除率降低。ADA对脑脊液Tofersen清除率的影响尚不清楚。未观察到ADA对总SOD1蛋白减少或血浆NfL减少的明显影响。未观察到 ADA 对安全性的明显影响。
【非临床毒理学】
尚未对Tofersen的致癌潜力进行评估;无致突变;高剂量下具有生育损害。
【临床研究】
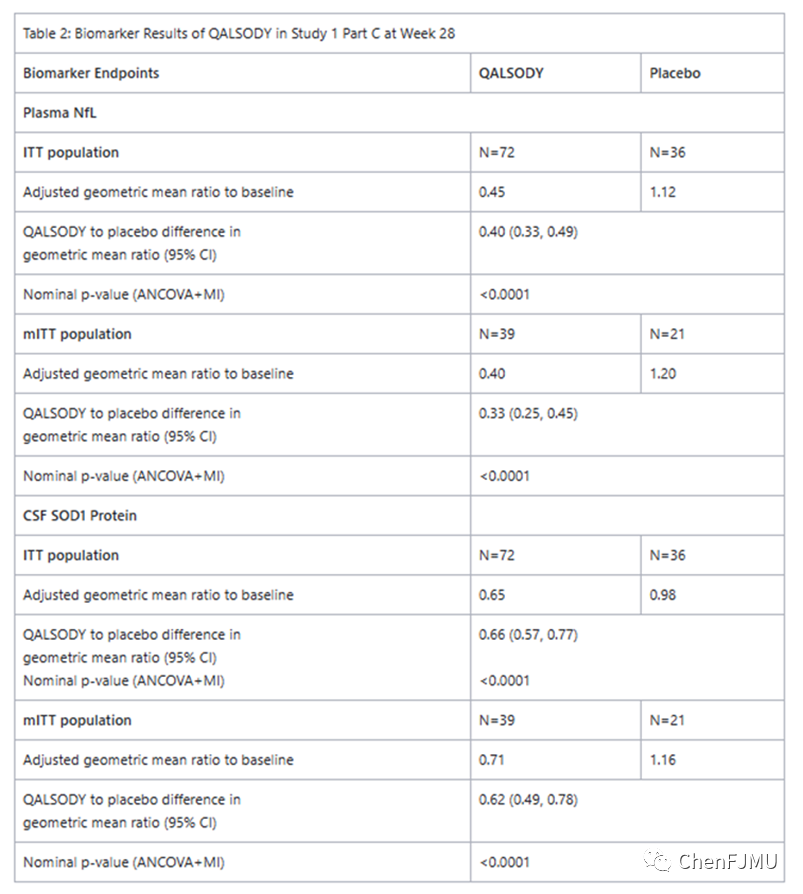
Tofersen的疗效是在一项随机、双盲、安慰剂对照的临床研究中评估(NCT02623699)。研究对象是23-78岁的、由SOD1突变引起的ALS患者。108名患者按2:1随机分组,接受Tofersen或安慰剂治疗,疗程为24 wk(3次负荷剂量,随后5次维持剂量)。Tofersen治疗组的平均基线ALSFRS-R评分为36.9(5.9)(ALSFRS-R评分是一种评估ALS患者疾病进展的常用工具,分数越低表示疾病进展越严重),安慰剂组为37.3(5.8)。Tofersen治疗组从症状发作的中位时间为11.4个月,安慰剂组为14.6个月。血浆NfL和脑脊液SOD1蛋白含量在第28 wk与基线相比变化具有统计学意义(见下表)。结果表明Tofersen治疗能使血浆NfL和脑脊液SOD1蛋白含量下降,对治疗ALS具有积极的疗效。
【用药指南】

Rilutek,02
Rilutek (Riluzole,利鲁唑)片剂由Sanofi公司最早进行研发,于1996年获FDA批准上市,是第一个获FDA批准用于治疗ALS的药物。Riluzole是一种新型谷氨酸调节剂,通过抑制电压依赖性Na+通道、高电压激活的Ca2+和K+通道和蛋白激酶C等复杂的机制,降低谷氨酸水平减少运动神经元的损伤,达到治疗目的。


Radicava,03
Radicava (Edaravone,依达拉奉)由日本三菱制药公司(Mitsubishi Tanabe Pharma Corporation)研发,于2017年5月5日获FDA批准上市,是一种自由基清除剂,能够缓解氧化应激的影响(这可能是ALS发病以及病情进展的关键因素),是FDA继Riluzole之后,批准的第二个治疗ALS的处方药物。
Relyvrio,04
Relyvrio(通用名:Sodium phenylbutyrate/Taurursodiol,苯丁酸钠/牛磺酸二醇复方制剂)由Amylyx公司研发,于2022年9月29日FDA批准上市,用于治疗肌萎缩侧索硬化成人患者,是FDA批准的第三款ALS处方药物。它们可以改善细胞内线粒体和内质网的健康状态,从而延缓神经细胞的死亡。


二、治疗脊髓性肌萎缩症SMA药物新盘点(ChenFJMU)
Spinraza
Spinraza(Nusinersen,诺西那生钠)由Ionis公司研发,是全球首款SMA治疗药物,于2016年12月23日获FDA批准上市,是一种针对运动神经元存活2(SMN2)的重磅反义寡核苷酸药物(ASO),在2022年全球销售额药物排行榜排名第152,销售额为17.93亿美元。旨在治疗由5q染色体突变引起的SMN蛋白缺乏导致的脊髓性肌萎缩症(SMA)罕见病。
SMA是一种遗传性神经肌肉疾病,主要影响脊髓神经元,导致肌肉萎缩和功能障碍。主要分为5个类型,分别为:O型(最严重)、I、 II、 III型脊肌萎缩症,这四种类型脊髓性肌肉萎缩症的症状首发于婴儿和儿童期;IV型脊髓性肌萎缩症在成年期首次发病。
Zolgensma(通用名:Onasemnogene abeparvovec,索伐瑞韦)是第二个获得FDA批准用于治疗SMA的治疗手段, 2019年5月在美国上市,属于基因替代疗法的一种。Evrysdi(Risdiplam,利司扑兰)是第三个获得FDA批准用于2月龄及以上儿童治疗SMA的药物,2019年5月在美国上市,属于小分子药物。
【结构与剂型处方】
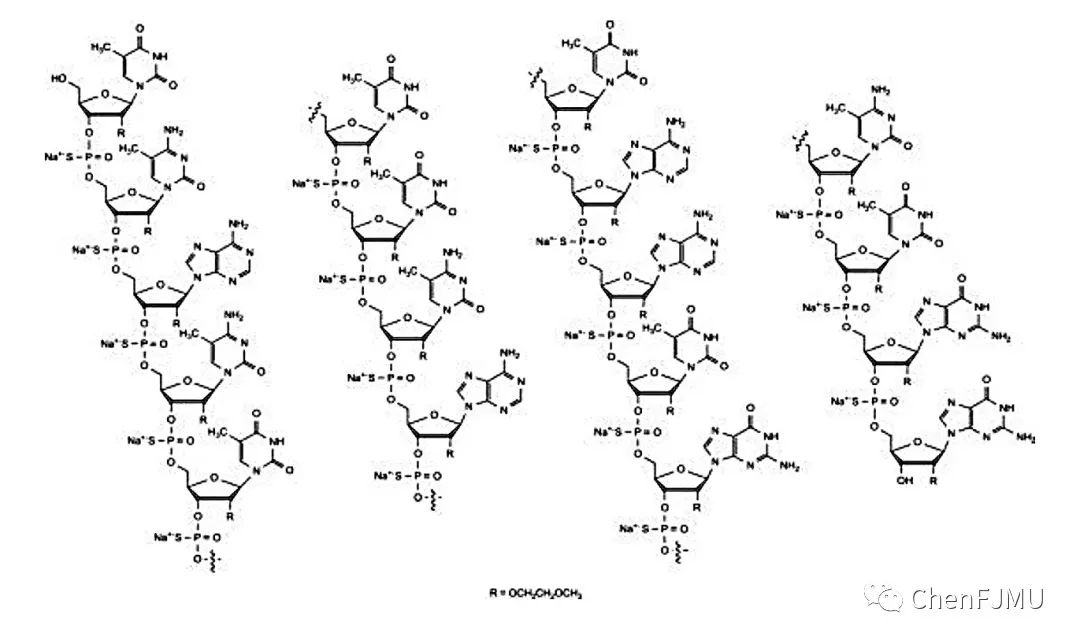
Nusinersen是一种修饰的反义寡核苷酸,序列为:5'-TCACTTTCATAATGCTGG-3'。17个硫代磷酸酯键PS,所有胞嘧啶碱基的5’引入了甲基(可消减免疫刺激副作用),糖环的2'-羟基被2'-O-甲氧基乙基(2'-O-MOE)取代。分子式C234H323N61O128P17S17Na17,分子量为7501.0。Nusinersen以注射剂的形式提供,用于单剂量玻璃小瓶鞘内使用。每1 mL 溶液含有2.4 mg Nusinersen, 还含有CaCl2·2H2O(0.21 mg)、MgCl2·6H2O(0.16 mg)、KCl(0.22 mg)、NaCl(8.77 mg)、无水磷酸二钠(0.10 mg)、磷酸二水合钠 (0.05 mg)和注射用水。Nusinersen含有盐酸或氢氧化钠以调节pH值(~7.2)。
【作用机制】
SMA是因为染色体5q13区域的SMN-1基因发生突变,导致运动神经元存活蛋白SMN表现不足。而SMN-2基因与SMN-1基因在外显子(exon) 7的碱基序列不同,SMN-2基因转录的mRNA缺乏外显子7(exon 7),转译出的SMN蛋白在功能上虽与SMN-1类似,但是功能性较差,而且不稳定,在体内容易被分解。Nusinersen是被设计为具有SMN-2碱基序列成互补关系的一种反义寡核苷酸,能与exon 7的剪切位点结合,改变SMN-2 premRNA剪切,转录出具有exon 7的mRNA,借此使SMN-2亦能制造完整长度的SMN蛋白,达到治疗的效果。
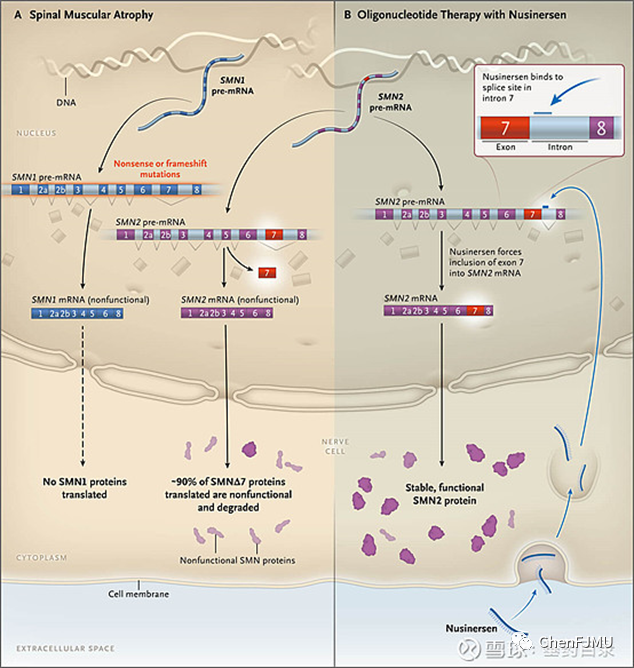
Crooke, S.T., et al. Nat Rev Drug Discov 20, 427–453 (2021).
【药效学及药代动力学】
药效学:与未经治疗的SMA婴儿相比,患者胸脊髓中含有更高水平的含有exon 7的SMN2 mRNA。接受治疗的患者QTcF值>2 ms,基线值>4 ms的变化,与延迟心室复极相关的心脏不良反应发生率没有增加。
吸收:Nusinersen经鞘内给药后进入脑脊液,并从脑脊液分布到中枢神经系统组织。与谷脑脊液浓度相比,Nusinersen的谷血浆浓度相对较低(谷浓度是指多次给药达稳态时给药后初始时刻至下次给药前的最低浓度)。中位血浆 Tmax值为1.7-6.0 h。平均血浆Cmax、AUC值增加至12 mg。
分布:Nusinersen分布在中枢神经系统和周围组织,如骨骼肌、肝脏和肾脏。
消除:Nusinersen通过核酸外切酶(3'-和5'-)水解代谢,与CYP450酶无关。在脑脊液中消除半衰期135-177 d,在血浆中为63-87 d。主要以原型或短链代谢物经尿液排出。
【非临床毒理学】
尚未对Nusinersen的致癌潜力进行评估,无遗传毒性,无生育损害。
【临床研究】
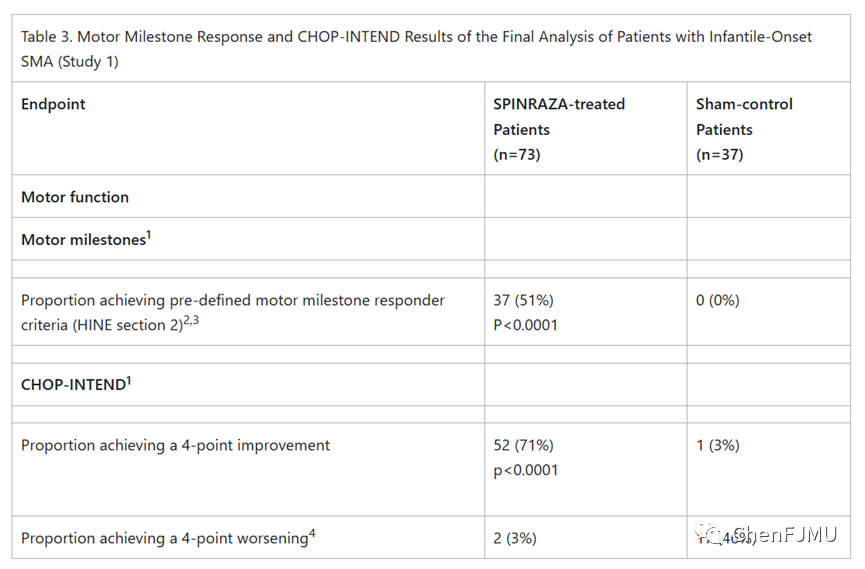
研究1 (NCT02193074)是一项多中心、随机、双盲、假手术对照研究,患者为首次给药时年龄≤7月龄、诊断为SMA(6月龄前出现症状)的有症状婴儿。在这项研究中,患者被认为最有可能发展为Ⅰ型SMA。中期分析时评估的主要终点是应答者的比例(根据Hammersmith婴儿神经检查(HINE)第2节标准来判断)。患者需要在更多的运动领域类别中表现出改善才可被评为应答者。在符合中期评估患者中,Nusinersen组中应答者的比例显著性提高,51%的患者达到了运动里程碑反应的定义,而假对照组中为0%。
研究2 (NCT02292537)是一项多中心、随机、双盲、假程序对照研究,患者为有症状的迟发性SMA儿童(6月龄后出现症状)。在这项研究中,患者被认为最有可能发展为Ⅱ型或Ⅲ型SMA。评估的主要终点是在第15个月时HFMSE的基线评分的变化。HFMSE评估行动受限的SMA患者的运动功能,得分越高表明运动功能越好。与假对照组相比,Nusinersen治疗组的HFMSE评分从基线到第15个月有显著改善。
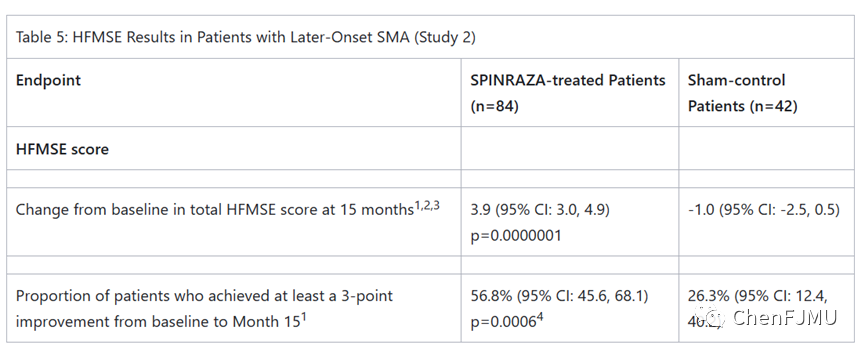
研究3(NCT02386553)在症状前患者中研究,这些患者遗传诊断为5q SMA和2或3个SMN2拷贝。根据WHO的运动里程碑对患者进行评估,所有在SMA症状发作前接受Nusinersen治疗的患者都存活了下来,不需要永久性通气,并且超出了基于其SMN2拷贝的预期。所有患者都达到了WHO的运动里程碑,即无需辅助就坐,88%患者达到了辅助行走。三项研究结果表明Nusinersen在SMA患者中有效,并且支持早期使用Nusinersen治疗。
【用药指南】

三、治疗杜氏肌营养不良DMD药物新盘点(ChenFJMU)
Exondys 51,01
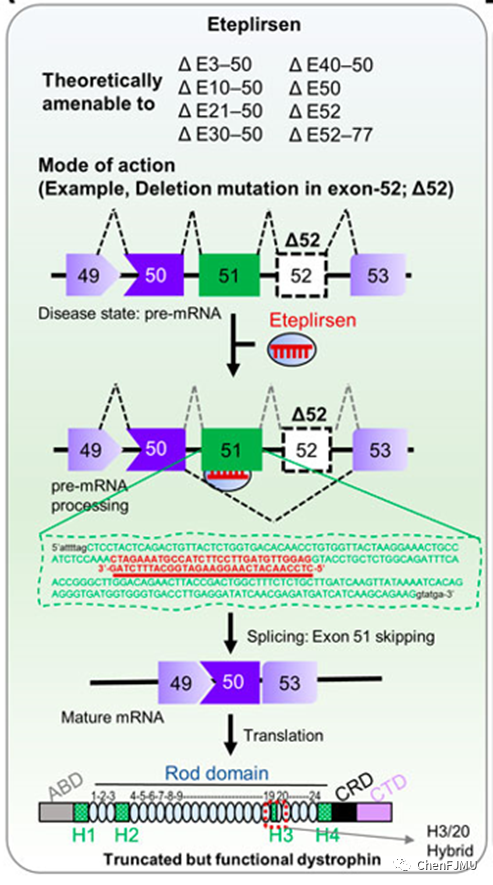
Exondys 51(Eteplirsen,依特普森)由Sarepta Therapeutics公司研发,于2016年9月19日获得美国FDA 加速批准上市。Eteplirsen是一种反义寡核苷酸药物(ASO,有争议),采用磷酰二胺吗啉代寡核苷酸(PMO)和外显子跳跃技术,适用于治疗确诊抗肌萎缩蛋白基因(DMD基因)突变且适合外显子51(exon-51)跳跃的杜氏肌营养不良(DMD)患者(约13%),可修复mRNA的阅读框来部分纠正遗传缺陷,成为首个获批治疗 DMD的药物。
DMD 是一种 X 染色体隐性遗传的进行性疾病,常伴有严重的并发症,包括心脏疾病或呼吸系统相关疾病。该病多见于男孩,一般在3-5岁开始发病,由DMD基因突变导致肌钙蛋白Dystrophin蛋白缺乏或异常而引起的肌肉萎缩性疾病。Dystrophin蛋白是稳定肌肉细胞膜的一个重要部份。它最重要的功能是维持肌肉细胞的稳定性,使它在肌肉收缩的过程中,不会受到破坏,DMD患者因肌肉中缺少了Dystrophin蛋白,使肌肉不断受到破坏和萎缩。DMD基因中有 79 个外显子,DMD 患者的外显子突变主要集中于外显子 45-55 的突变。由于 DMD 基因移码突变,导致生成的mRNA不能编码正常工作的Dystrophin蛋白。
Eteplirsen上市后饱受争议:用替代终点来加速通过上市批准,将肌肉dystrophin蛋白含量作为一个终点,替代了DMD治疗的主要终点(NSAA、6分钟步行等行动指标)。临床试验设计存在严重缺陷:201/202临床试验中仅有12名患者,样本不足;且FDA测出的dystrophin蛋白含量与Serepta先前数据不一致。补充提交301试验数据,dystrophin蛋白含量也仅提高了0.28%。
【结构与剂型处方】
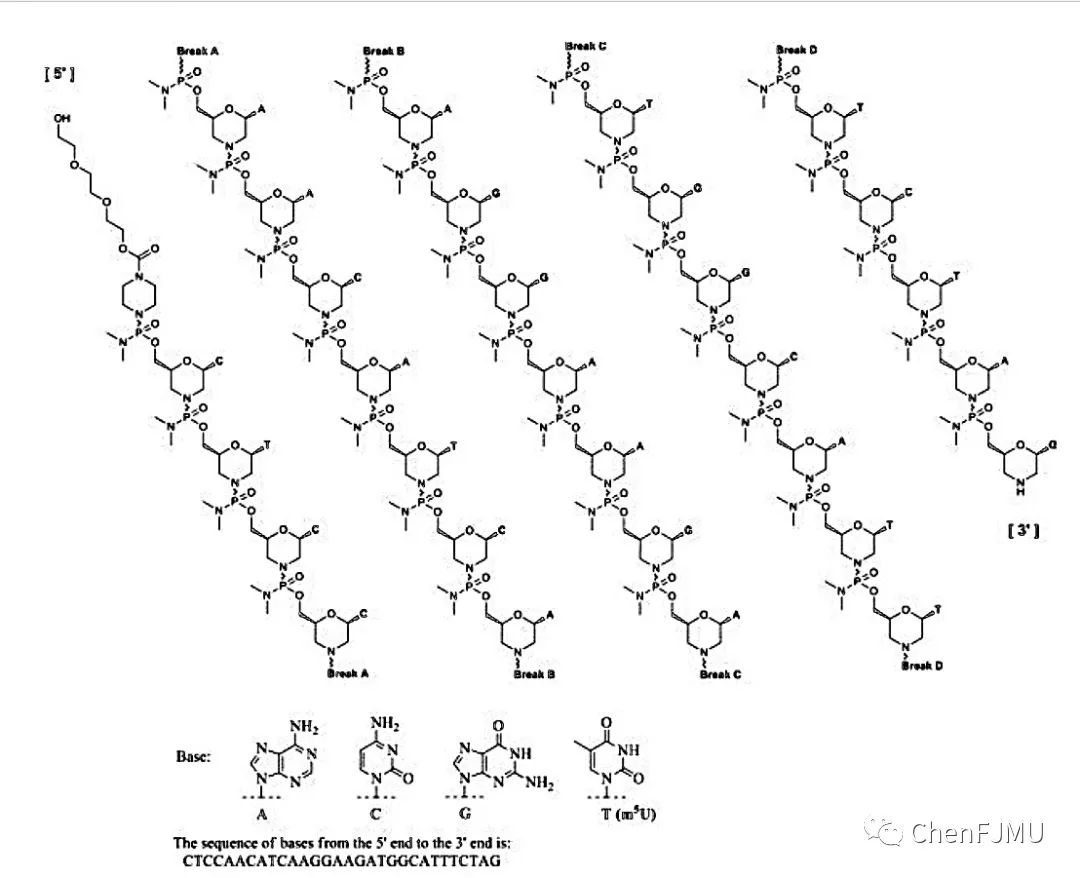
Eteplirsen是磷酰二胺吗啡啉寡核苷酸(吗啉代寡核苷酸,PMO),序列为5’-CTCCAACATCAAGGAAGATGGCATTTCTAG-3’。PMO是一类具有独特结构的ASO,结构中以吗啉环代替DNA或RNA糖环结构,以磷酰二胺酯键作为桥连骨架。电中性的吗啉-磷酰二胺结构使其具有靶RNA结合亲和性高、抗酶解稳定性强的特点(体内半衰期延长),减少与蛋白质的不利相互作用,不会激活宿主免疫应答,毒性较小。PMO结构特点决定了它不会引起靶RNA的降解,但可与靶RNA稳定、持续的序列特异性结合,使其能以立体位阻的方式,干扰靶RNA的下游活动:前信使RNA (pre-mRNA)剪切、mRNA翻译、miRNA加工,导致靶蛋白合成被阻断、产生新功能蛋白,达到在RNA水平矫正蛋白质功能的目的。
Eteplirsen含有30个碱基,分子式为C364H569N177O122P30,分子量为10305.7。Eteplirsen以注射剂的形式提供,每毫升含有50 mg Eteplirsen,0.2 mg KCl,0.2 mg KH2PO4,8 mg NaCl,1.14 mg Na2H2P2O7,注射用水。本产品可能含有盐酸或氢氧化钠来调节pH值。
【作用机制】
Eteplirsen是外显子跳跃技术的ASO,DMD患者的基因突变会使得片段丢失或者扩增,造成阅读框移动,导致抗肌萎缩蛋白的合成过早中断,生成的蛋白无法与肌肉细胞中的其他蛋白结合,丧失其正常功能。外显子跳跃技术的原理是通过改变表达抗肌萎缩蛋白的pre-mRNA的剪接过程,去除了基因突变带来的阅读框移动,跳过让蛋白合成过早中断的基因变异,生成的抗肌萎缩蛋白虽然比正常蛋白要小一些,但是保留了大部分与其他蛋白结合的结构域,仍然可以行使蛋白的部分正常功能。Eteplirsen与肌钙蛋白pre-mRNA exon-51结合,使得exon-51跳跃,恢复阅读框,产生截短但有功能性蛋白。
打个比喻:
正常人Dystrophin蛋白:
The mad cat ate the fat rat.
BMD病人Dystrophin基因突变:
The mad cat ate the fat rat.
The mad cat ate the rat.
DMD病人Dystrophin基因突变:
The mad cat ate the fat rat.
The mad cat ate hef atr at.
DMD病人采用exon skipping ASO疗法:
The mad cat ate the fat rat.
Aoki, Front. Genome Ed. 2022
【药效学及药代动力学】
药效学:所有接受Eteplirsen治疗患者均通过RT-PCR反应产生截短Dystrophin的mRNA。Eteplirsen治疗后,患者肌肉组织中Dystrophin的平均水平增加。
吸收:患者接受Eteplirsen治疗后,血药浓度-时间谱大致相似,且呈下降趋势。大部分药物消除发生在24 h内。每周给药后没有明显的药物积累。受试者间Cmax和AUC的变化范围为20%-55%。单次或多次静脉输注Eteplirsen的峰值血浆浓度(Cmax)发生在输注结束时。
分布:Eteplirsen在人体内的血浆蛋白结合率在6-17%。每周静脉输注后,其平均表观分布体积(Vss)为600 mL/kg。注射结束24 h后,平均浓度为Cmax的0.07%。在每周一次给药期间,没有观察到药物积累。
消除:用339 mg/kg/wk治疗12 wk后,Eteplirsen的总清除率为30 mL/hr/kg。
Eteplirsen不被任何物种的肝脏微粒体代谢。Eteplirsen的肾脏清除率约占静脉给药后24 h内给药剂量的三分之二。消除半衰期(t1/2)是3-4 h。
特殊人群:与肾功能正常的受试者相比,轻度和中度肾损伤的受试者表现出更高的Eteplirsen暴露,暴露量(AUC)分别增加了约1.4倍和2.4倍。严重肾损伤或终末期肾病对Eteplirsen药代动力学和安全性的影响尚未研究。对于肾功能损害患者,不建议进行特定的剂量调整。Eteplirsen尚未在肝损伤患者中进行研究。
药物相互作用研究:Eteplirsen对CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6和CYP3A4/5均无明显抑制作用。不能诱导CYP2B6或CYP3A4,对CYP1A2的诱导小于奥美拉唑诱导剂。Eteplirsen不是底物,也不是转运蛋白抑制剂(OAT1、OAT3、OCT1、OCT2、OATP1B1、OATP1B3、P-gp、BCRP、MRP2和BSEP)。
【非临床毒理学】
尚未对Eteplirsen的生育损害进行评估;无诱变;无致癌。
【临床研究】
在研究1中,患者随机接受每周输注Eteplirsen (30 mg/kg, n=4); Eteplirsen (50 mg/kg, n=4)或安慰剂(n=4)治疗24 wk。主要终点是Dystrophin蛋白的产生,同时评估6 min步行测试(6MWT)。6MWT测量患者在6 min内在平坦坚硬的表面上行走的距离。患者基线时平均6 min步行距离(6MWD)为363 m,并且使用稳定剂量的皮质类固醇至少6个月。接受Eteplirsen治疗的患者与接受安慰剂治疗的患者之间的6MWD变化无显著差异。所有参加研究1的12名患者在研究2中继续接受开放标签Eteplirsen每周治疗4年。4名被随机分配到安慰剂组的患者以1:1的比例重新随机分配到Eteplirsen(30或50 mg/kg/wk)治疗,使得每个剂量有6名患者。参与研究2的患者与外部对照组进行比较。主要临床疗效指标为6MWT。
研究2中的11例患者在接受Eteplirsen治疗180 wk后进行了肌肉活检,并通过WB分析了肌营养不良蛋白水平。与外部对照组相比,研究2未能提供Eteplirsen临床获益的证据。用Eteplirsen治疗180 wk后,平均肌营养不良蛋白水平为健康受试者的0.93%。由于研究1中使用Eteplirsen治疗前的肌营养不良蛋白水平信息不足,因此无法估计研究1中Eteplirsen对肌营养不良蛋白产生的反应。
在研究3中,13名患者每周接受开放标签Eteplirsen (30 mg/kg)治疗48 wk,并在基线和治疗48 wk后进行肌肉活检。患者服用稳定剂量的皮质类固醇至少6个月。WB检测肌肉组织中肌营养不良蛋白水平。在12例可评价结果的患者中,治疗前的肌营养不良蛋白水平为健康受试者的0.16%±0.12%,Eteplirsen治疗48 wk后为0.44%±0.43% (p < 0.05)。48 wk后的平均增幅为0.28%。以上的数据都不能提供明显的证据来证明Eteplirsen可显著提高患者Dystrophin含量,因此使其备受争议。
【用药指南】

Vyondys 53,02
Vyondys 53(Golodirsen,戈洛迪森)是由Sarepta Therapeutics公司研发的第二款治疗DMD的反义寡核苷酸药物,经美国FDA加速批准于2019年12月12日上市。其采用了磷酰二胺吗啡啉寡核苷酸PMO和外显子跳跃技术,适用于治疗确诊DMD基因突变且适合外显子53(exon-53)跳跃的DMD患者(约8%),pre-mRNA exon-53结合,exon-53跳跃,修复mRNA阅读框来部分纠正遗传缺陷,产生截短但有功能Dystrophin蛋白。
【结构与剂型处方】
Golodirsen是磷酰二胺吗啡啉寡核苷酸(吗啉代寡核苷酸,PMO),序列为:5’-GTTGCCTCCGGTTCTGAAGGTGTTC-3’。Golodirsen结构中以吗啉环代替糖环结构,以磷酰二胺酯键作为桥连骨架。Golodirsen结构呈电中性,使其具有与靶RNA亲和性高、抗酶解稳定性强(体内半衰期延长)等特点。Golodirsen含有25个碱基,分子式为C305H481N138O112P25,分子量为8647.28。以注射剂形式提供,配制成等渗磷酸盐缓冲盐溶液,渗透压为260-320 mOsm,pH值为7.5。每毫升含有50 mg Golodirsen,0.2 mg KCl,0.2 mg KH2PO4,8 mg NaCl,1.14 mg Na2H2P2O7,注射用水。本产品含有盐酸或氢氧化钠来调节pH值。

【作用机制】
Golodirsen是外显子跳跃技术ASO,通过改变表达抗肌萎缩蛋白的mRNA的剪接过程,与Dystrophin pre-mRNA exon-53结合,使得exon-53跳跃,恢复阅读框,跳过让蛋白合成过早中断的基因突变,产生截短但有功能性蛋白。
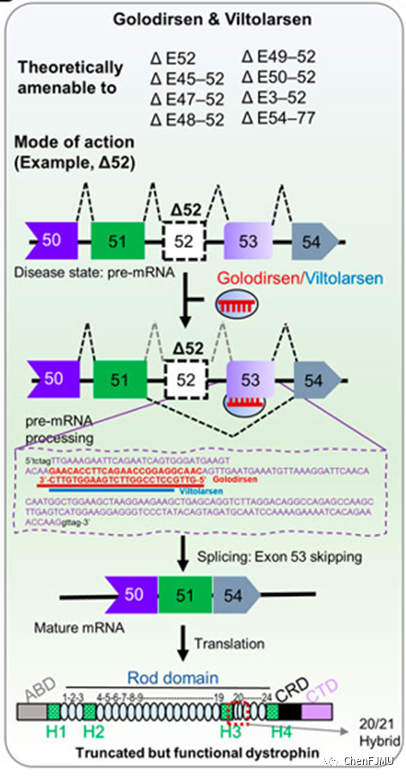
Aoki, Front. Genome Ed. 2022
【药效学及药代动力学】
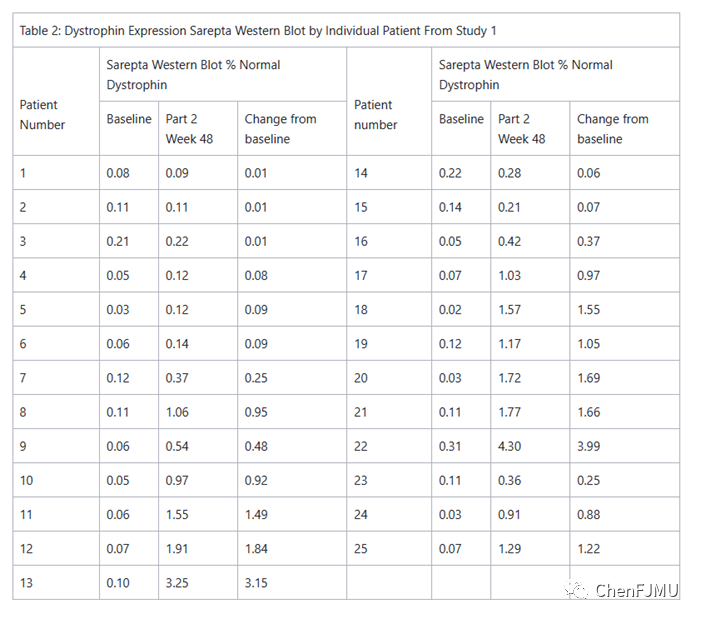
药效学:用Golodirsen治疗后,与基线相比,患者通过RT-PCR证明exon-53的跳跃增加。治疗48 wk后,用WB检测的肌营养不良蛋白水平从基线时正常水平的0.10%上升到1.02%,肌营养不良蛋白与基线相比的平均变化为正常水平的0.92%。这种肌营养不良蛋白表达的增加与外显子跳变水平呈正相关。免疫荧光染色证实了Golodirsen治疗患者肌纤维中截断的肌营养不良蛋白在肌膜上的正确定位。
吸收:Golodirsen暴露量随剂量成比例增加,每周给药一次积累最小。Cmax和AUC的受试者间差异(以%CV表示)分别为38%-72%和34%-44%。
分布:稳态体积分布在DMD患者和健康受试者之间相似。在剂量为30 mg/kg时,平均分布体积为668 mL/kg (%CV=32.3)。Golodirsen血浆蛋白结合范围为33%-39%,不依赖于浓度。
消除:Golodirsen消除半衰期为3.4 h,在346 mg/kg剂量下血浆清除率为30 mL/hr/kg。Golodirsen代谢稳定,在血浆或尿液中未检测到代谢物。大部分随尿液排出。
特定人群:41-65岁的非DMD患者中评估了肾损伤对Golodirsen药代动力学的影响。在患有2期或3期CKD的受试者中,暴露(AUC)分别增加了约1.2倍和1.9倍。与肾功能正常的受试者相比,患有2期CKD的受试者中Cmax无变化;患有3期CKD的受试者中Cmax增加了1.2倍。不建议肾功能损害患者进行特定剂量调整。尚未在肝损伤患者中进行研究。
药物相互作用:Golodirsen对CYP1A2、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6和CYP3A4/5均无体外抑制作用,是CYP1A2的弱诱导剂,对CYP2B6和CYP3A4没有诱导作用。不被人肝微粒体代谢,也不是药物转运体(OAT1、OAT3、OCT2、OATP1B1、MATE1、P-gp、BCRP和MRP2、OATP1B3和MATE2-K)的底物或强抑制剂。
【非临床毒理学】
尚未对Golodirsen的致癌潜力进行评估;无致突变;无生育损害。
【临床研究】
在一项针对DMD患者的研究中评估了Golodirsen对肌营养不良蛋白产生的影响,这些患者确认DMD基因突变适合外显子53跳跃(研究1;NCT02310906)。研究1第1部分是一项双盲、安慰剂对照、剂量递增研究,共12名DMD患者,8名患者接受Golodirsen治疗,4名接受安慰剂治疗。Golodirsen治疗的患者按照4个逐步递增的剂量水平,从4 mg/kg/wk-30 mg/kg/wk,在每个剂量水平静脉输注2 wk。研究1第2部分是一项为期168 wk的开放标签研究,包括第1部分的12名患者和13名新的DMD患者。评估了Golodirsen疗效和安全性,剂量为30 mg/kg/wk,在研究开始时服用稳定剂量的皮质类固醇至少6个月。根据第2部分第48 wk时肌营养不良蛋白水平与基线的变化来评估疗效。采用肌肉活检,以及WB来分析肌营养不良蛋白水平。平均肌营养不良蛋白水平从基线时正常水平的0.10%上升到正常水平的1.02%,其中平均变化为正常水平的0.92%;与基线相比,中位数变化为0.88%,表明有疗效。
【用药指南】

Viltolarsen,03
Viltepso(Viltolarsen,维托拉森)由NS Pharma 公司研发,于2020年8月12日获美FDA批准上市,是第三款治疗DMD的ASO药物。Viltolarsen也是一种磷酰二胺吗啡啉寡核苷酸PMO,靶向外显子53(exon-53)与Golodirsen相同区域。然而,Viltolarsen是21-mer,而Golodirsen是25-mer寡核苷酸。Viltolarsen与Golodirsen一样采用了PMO和外显子跳跃技术,适用于治疗确诊DMD基因突变且适合exon-53跳跃的DMD患者(约8%),与pre-mRNA exon-53结合,exon-53跳跃,修复mRNA的阅读框来部分纠正遗传缺陷,产生截短但有部分功能的Dystrophin蛋白。
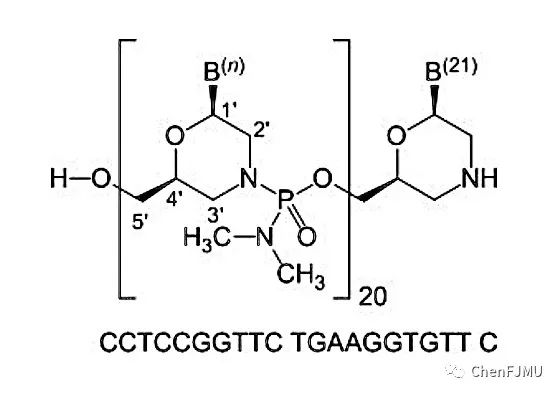
【结构与剂型处方】
Viltolarsen是磷酰二胺吗啡啉寡核苷酸(吗啉代寡核苷酸,PMO),序列为21-mer:5’-CCTCCGGTTCTGAAGGTGTTC-3’;而Golodirsen序列为25-mer:5’-GTTGCCTCCGGTTCTGAAGGTGTTC-3’。Viltolarsen结构以吗啉环代替糖环结构,以磷酰二胺酯键作为桥连骨架,呈电中性,使其具有与靶RNA亲和性高、抗酶解稳定性强等特点。分子式为C244H381N113O88P20,分子量为6924.82。Viltolarsen以注射剂形式提供,每毫升含有50 mg的Viltolarsen和9 mg NaCl。使用盐酸或氢氧化钠将产品调节至7.0-7.5之间的pH值。
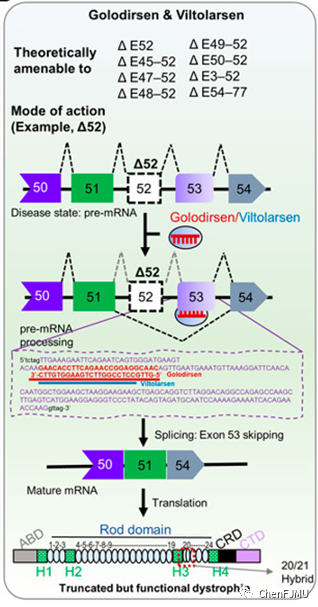
【作用机制】
Viltolarsen是外显子跳跃技术ASO,与Golodirsen作用机制相同,通过改变表达抗肌萎缩蛋白的mRNA的剪接过程,与其pre-mRNA exon-53结合,使得exon-53跳跃,恢复阅读框,跳过让蛋白合成过早中断的基因突变,产生截短但有功能Dystrophin蛋白。
Aoki, Front. Genome Ed. 2022
【药效学及药代动力学】
药效学:接受Viltolarsen治疗后,通过RT-PCR检测,所有患者均产生一种截断的肌营养不良蛋白mRNA,并通过DNA序列分析显示exon-53跳跃。通过WB方法量化,均显示肌营养不良蛋白表达较基线增加。免疫荧光染色证实了维托拉森治疗患者的肌纤维中肌营养不良蛋白的截短定位。
药代动力学:在静脉输注剂量为1.25 mg/kg/wk-80 mg/kg/wk的DMD患者中,对Viltolarsen的药代动力学进行了评估。Viltolarsen暴露量随剂量成比例增加,每周给药一次积累最小。Cmax和AUC的受试者间变异性(以%CV表示)分别为16%-27%。
分布:在剂量为80 mg/kg时,Viltolarsen的平均稳态分布体积为300 mL/kg (%CV=14)。Viltolarsen血浆蛋白结合范围为39%-40%,不依赖于浓度。
消除:Viltolarsen代谢稳定,血浆和尿液中未检测到代谢物。除半衰期为2.5 h,血浆清除率为217 mL/hr/kg。
特定人群:Viltolarsen尚未在肾脏或肝脏损伤患者中进行研究。
药物相互作用:Viltolarsen对CYP3A4/5、CYP1A2、CYP2A6、CYP2B6、CYP2C8、CYP2C9、CYP2C19、CYP2D6、CYP2E1、UGT1A1和UGT2B7均无抑制作用。不诱导CYP1A2、CYP2B6或CYP3A4。不被CYP酶代谢,也不是转运体BCRP、BSEP、MDR1、OAT1、OAT3、OCT1、OCT2、MATE1或MATE2-K的底物。对转运蛋白(OATP1B1、OATP1B3、OAT3、BCRP、MDR1、BSEP、OAT1、OCT1、OCT2、MATE1和MATE2-K)没有抑制作用。
免疫原性:与所有寡核苷酸一样,存在潜在的免疫原性。评估了在第16 d(给药前)、第 1、5、13 wk患者体内抗Viltolarsen抗体,结果为阴性。在第1、16 wk,13.24%患者检测到抗肌营养不良蛋白抗体;在第37、49、73、97 wk,抗体消失。此外,患者的肌营养不良蛋白水平与基线相比发生了变化,与他的剂量组的平均变化相当,并且该抗体产生没有报告不良事件。另一研究患者均被确定为抗Viltolarsen抗体和抗肌营养不良蛋白抗体阴性。Viltolarsen缺乏观察到的免疫原性,表明其免疫原性不高。
【非临床毒理学】
尚未对Viltolarsen的致癌潜力进行评估;无致突变;无生育损害。
【临床研究】
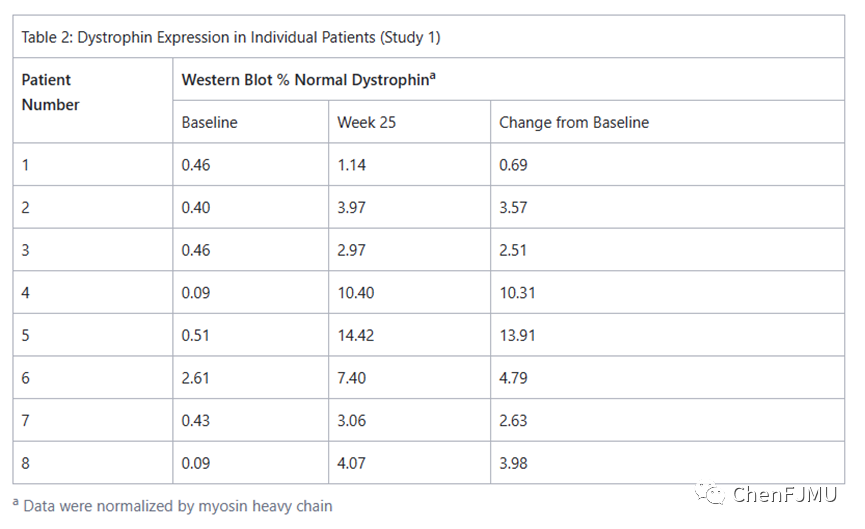
在一项针对DMD患者的研究中评估了Viltolarsen对肌营养不良蛋白产生的影响,这些患者确认DMD基因突变适合外显子53跳跃(研究1;NCT02740972)。在研究1的前4 wk,患者随机(双盲)接受Viltolarsen或安慰剂治疗。随后接受20 wk的开放标签治疗,40 mg /kg每周1次或80 mg /kg每周1次。患者接受稳定的皮质类固醇治疗至少3个月。根据第25 wk时肌营养不良蛋白水平的基线变化来评估疗效。在基线和Viltolarsen治疗24 wk后收集患者的肌肉活检,并通过标准化为肌球蛋白重链(主要终点)和质谱(次要终点)的蛋白质印迹分析肌萎缩蛋白水平。在每周一次接受 Viltolarsen的患者中,平均肌营养不良蛋白水平从基线时正常值的 0.6%增加到第5 wk的正常值的9.4%,通过验证的蛋白质印迹评估,肌营养不良蛋白的平均变化为正常水平的5.3%;与基线相比的中位变化为3.8%。所有患者的肌营养不良蛋白水平均高于基线值。通过质谱法(标准化为丝层蛋白C)评估,平均肌营养不良蛋白水平从基线时正常值的0.6%增加到第4 wk的正常值的2.3%,抗肌萎缩蛋白的平均变化为正常水平的3.7%;与基线相比的中位变化为1.9%,以上数据表明有疗效。
【用药指南】

Casimersen,04
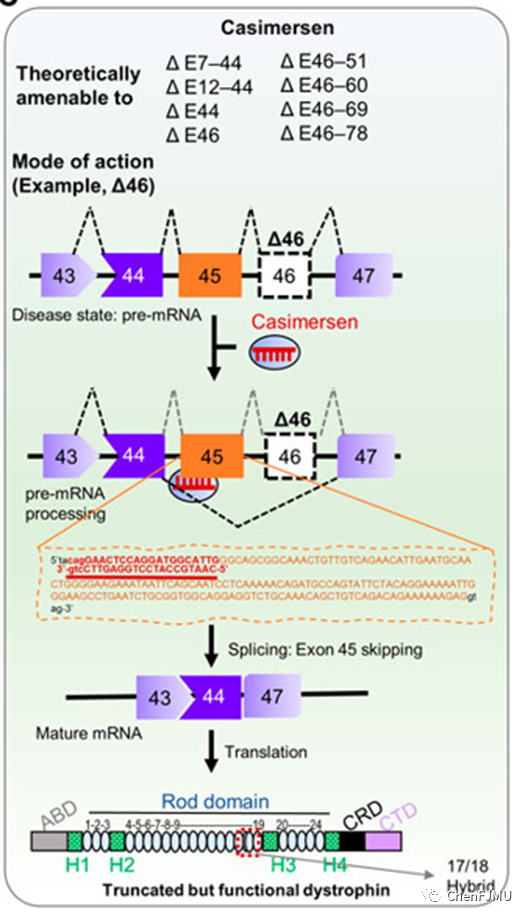
Amondys 45(Casimersen,卡西默森)由Sarepta Therapeutics公司研发,2021年2月15日获美FDA批准上市,Casimersen成为继Exondys 51(eteplirsen)、Vyondys 53(golodirsen)和Viltepso(Viltolarsen)之后,第4个在美国获批的外显子跳跃技术的反义寡核苷酸疗法。适用于治疗确诊DMD基因突变且适合外显子45(exon-45)跳跃的DMD患者(约8%),Casimersen 与pre-mRNA exon-45结合,使exon-45跳跃,修复mRNA的阅读框来部分纠正遗传缺陷,产生截短有功能Dystrophin蛋白。
【结构与剂型处方】
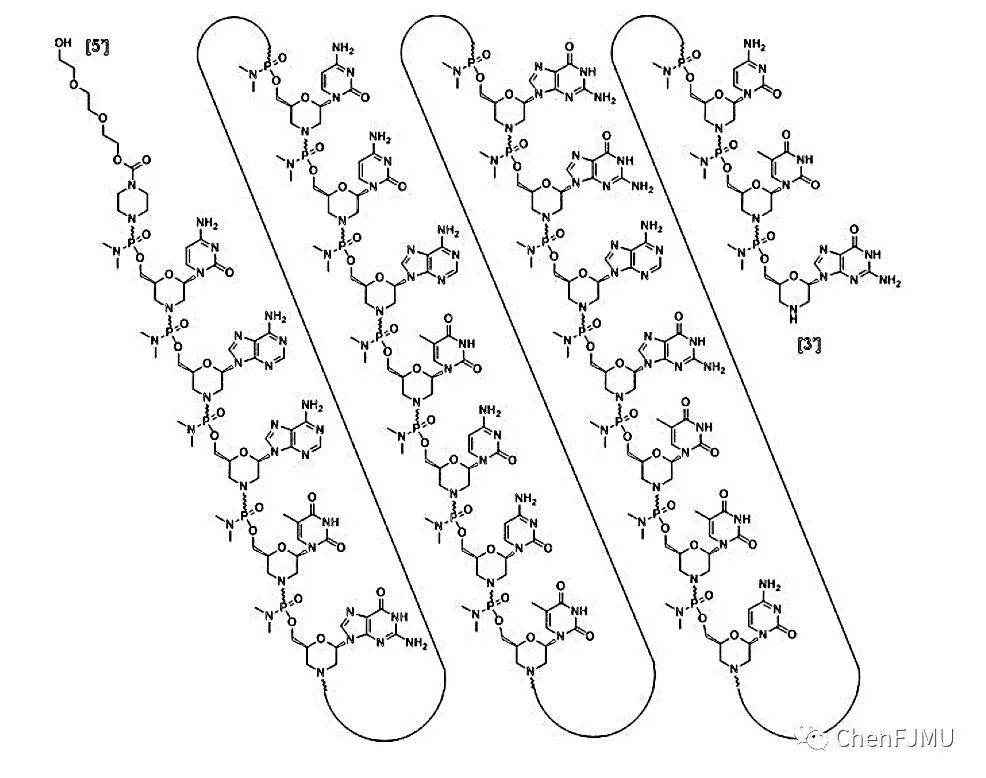
Casimersen是磷酰二胺吗啡啉寡核苷酸(吗啉代寡核苷酸,PMO),序列为5’-CAATGCCATCCTGGAGTTCCTG-3’。Casimersen结构以吗啉环代替糖环结构,以磷酰二胺酯键作为桥连骨架,呈电中性,使其具有与靶RNA亲和性高、抗酶解稳定性强等特点。Casimersen含有22个碱基,分子式为C268H424N124O95P22,分子量为7584.5。Eteplirsen以注射剂形式提供,配制成等渗磷酸盐缓冲盐溶液,渗透压为 260-320 mOsm,pH值为7.5。每毫升含有50 mg Casimersen;0.2 mg KCl,0.2 mg KH2PO4,8 mg NaCl和1.14 mg NaH2PO4,注射用水。该产品含有盐酸或氢氧化钠以调节pH值。
【作用机制】
Casimersen是外显子跳跃技术ASO,可通过改变表达抗肌萎缩蛋白的mRNA的剪接过程,与其pre-mRNA exon-45结合,使得exon-45跳跃,去除了基因突变带来的阅读框移动,恢复阅读框,跳过让蛋白合成过早中断的基因变异,产生截短Dystrophin蛋白。
Aoki, Front. Genome Ed. 2022
【药效学及药代动力学】
药效学:从患者的肌肉活检组织分析中,接受Casimersen的患者与基线相比,exon-45的跳跃显著增加,通过RT-PCR证明。外显子跳跃的水平与肌营养不良蛋白表达呈正相关。通过WB测定评估肌营养不良蛋白水平从基线时正常值的0.93%增加到正常值的1.97%(治疗74 wk后)。与接受安慰剂的患者相比,接受Casimersen的患者从基线到第48 wk的肌营养不良蛋白水平显著增加。免疫荧光染色证明,在接受Casimersen治疗的患者中,肌营养不良蛋白正确定位到肌膜。
药代动力学:单次静脉注射Casimersen后,Cmax在输液结束时达到。暴露量随剂量增加而成比例地增加。每周一次给药后,血浆中未观察到Casimersen的积累。Cmax和AUC在受试者间变异性分别为12% ~ 34%和16% ~ 34%。
分布:Casimersen与人血浆蛋白的结合不受浓度的影响,其结合率在8.4%-31.6%之间。静脉注射30 mg/kg后,稳态平均表观分布体积(Vss)为367 mL/kg (%CV = 28.9)。
消除:30 mg/kg剂量时,Casimersen的血浆清除率为180 mL/hr/kg。消除半衰期(t1/2)为3.5 h。Casimersen在人肝微粒体培养中代谢稳定。血浆和尿液中未检测到代谢物。90%以上的药物通过尿液排出。
特定人群:在35-65岁的2或3期慢性肾脏病(CKD)的非DMD受试者中评估肾损伤对Casimersen药代动力学的影响。受试者接受单次30 mg / kg静脉注射剂量的Casimersen。在患有2期或3期CKD的受试者中,与肾功能正常的受试者相比,暴露(AUC)分别增加了约1.2倍和1.8倍。2期CKD患者的Cmax与肾功能正常者相似;CKD 3期患者的Cmax增加了1.2倍。4期或5期CKD对Casimersen药代动力学和安全性的影响尚未研究。不建议对肾功能损害患者进行特定的剂量调整。Casimersen尚未在肝损伤患者中进行研究。
药物相互作用:Casimersen与CYP酶和转运体发生药物相互作用的可能性较低。Casimersen对CYP1A2、CYP2B6、CYP2C8和CYP2D6均无体外抑制作用。是体外CYP3A4/5、CYP2C9和CYP2C19的潜在抑制剂。Casimersen在mRNA或蛋白水平上均未诱导CYP1A2、CYP2B6或CYP3A4。被人肝微粒体代谢,不是药物转运体(OAT1、OAT3、OCT2、OATP1B1、OATP1B3、MATE1、MATE2-K、P-gp、BCRP和MRP2)的底物或强抑制剂。
【非临床毒理学】
尚未对Casimersen的致癌潜力进行评估;无致突变;无生育损害。
【临床研究】
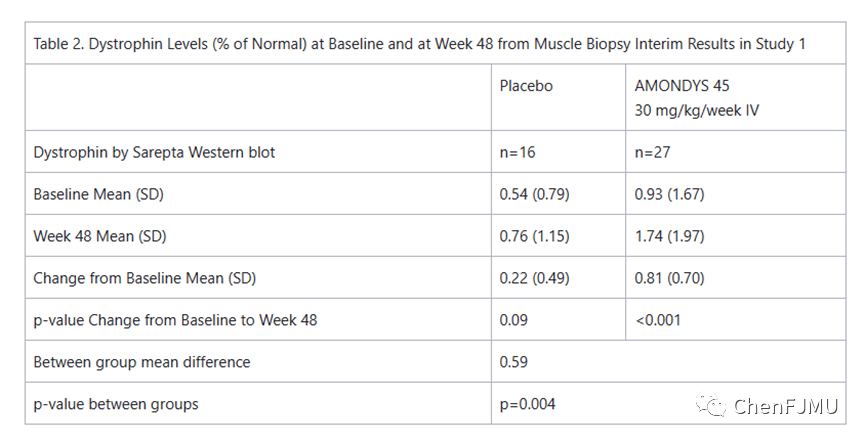
研究1(NCT02500381)是一项正在进行的双盲、安慰剂对照、多中心研究,旨在评估Casimersen在患者中的安全性和有效性。在服用Casimersen或安慰剂之前,患者需要服用稳定剂量的口服皮质类固醇至少24 wk。96 wk双盲期后,所有患者开始或即将开始额外的48 wk开放标签治疗期。中期疗效是根据研究1第48 wk时肌营养不良蛋白水平与基线的变化来评估的。在双盲期第48 wk进行肌肉活检的患者平均值为1.74%,与基线相比提高了0.81%。
【用药指南】

四、治疗弗里德赖希共济失调FRDA药物新盘点(ChenFJMU)
Omaveloxolone
Skyclarys(Omaveloxolone,奥马索龙)由Reata Pharm.公司研发,FDA于2023年2月28日批准,Skyclarys是用于治疗弗里德赖希共济失调FRDA的处方药,这是一种罕见的神经肌肉疾病,会导致力量和协调能力丧失,最终导致患者需乘坐轮椅。核因子- E2相关因子2 (Nuclearfactor erythroidderived 2-like 2, Nrf2)与氧化应激、线粒体功能障碍和细胞损伤有关,包括中枢和外周神经元。Omaveoxolone已证明可在动物和人的体内外激活Nrf2途径。Nrf2通路参与细胞对氧化应激的反应。FDA已授予Omaveloxolone孤儿药资格、快速通道资格、及罕见儿科疾病认定。
四种神经肌肉罕见病药物新盘点
(ChenFJMU)