罕见病治疗仍是一个全球性难题,普遍面临治疗药物有限,可及性较低的困境。让罕见病有药可治,是我国“提高罕见病救治水平”的重要目标,也是全球医学持续进步的意义所在。值此2023开年之际,采撷过去一两年间罕见病药物的研发进展,为2022画上句号,为新年度怀梦启航。
进行性肌营养不良
进行性肌营养不良是一组以骨骼肌进行性无力萎缩为主要临床表现的异质性基因缺陷性疾病。可伴有中枢神经系统、心脏、骨骼、呼吸及胃肠道受累。不同类型的起病时间、进展速度、严重程度的差异很大。其中Duchenne肌营养不良(DMD)由抗肌萎缩蛋白基因突变导致的抗肌萎缩蛋白缺失引起,全球范围内新生儿男婴的发病率约为1/3500。患者的DMD基因大片段缺失主要集中在3-22号和45-54号外显子区域,发生率约为60%;其次为点突出,发生率30%。
通过规范药物治疗、康复训练、定期随诊评估相关系统受累并给予治疗,能够明显延缓DMD疾病进展,延长生存期,只是目前该病仍无法治愈。常规药物治疗主要指早期、规范地给予口服激素药物,如泼尼松、泼尼松龙、甲泼尼龙、地夫可特,激素可抑制机体的免疫功能,抑制生长激素分泌影响儿童生长发育,是医患痛点,亟需新疗法的出现。
新药物.新实践
2016-2021年,FDA先后批准Sarepta公司的三款外显子跳跃疗法,分别是用于51号外显子跳跃缺失DMD的Eteplirsen(Exondys51)、外显子53跳跃的Golodirsen(Vyondys 53)、外显子45跳跃的Casimersen (Amondys 45)。2020年,还有来自日本新药株式会社的Viltolarsen(Viltepso),作为第二款外显子53跳跃的药物获FDA批准。这四款均属于磷酰二胺吗啉代低聚物反义寡核苷酸(ASO)药物,可部分恢复致病基因的阅读框,以增加患者抗肌萎缩蛋白水平。ASO类药物是新型DMD治疗手段,它们的上市共同开启了基因治疗、细胞治疗新时代。 表. 国际上已批准上市用于治疗DMD的4种ASO药物 (许婷婷,等.罕见病研究[J],2022)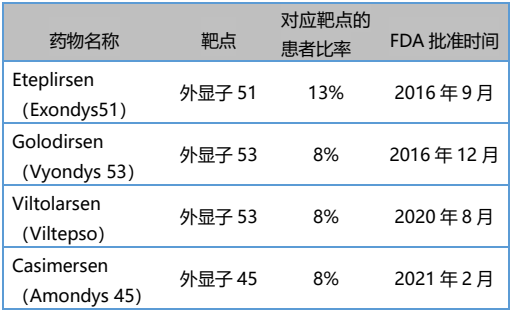
不远的将来,这些新型药物有望通过不同途径为中国DMD患者所用。如,Sarepta Therapeutics公司的三款DMD药物,自2021年即由博鳌乐城维健罕见病临床医学中心引入国内先行使用。对于Viltolarsen,国家药品监督管理局药品评审中心(CDE)官网显示其以“维托拉生注射液”中文名获得两项临床试验的默示许可,治疗已确认DMD基因突变且适合外显子53跳跃治疗的DMD患者;并于2022年6月,进口申请获CDE受理。2022年7月,国际期刊J Neuromuscul Dis报告了维托拉生的开放标签、长期扩展研究截至109周时的结果,在主要研究终点由仰卧至站立所需时间(TTSTAND)的评估上,可见第73周、109周明显改善;在次要研究终点10米移动的测试(TTRW)的评估上,结论相同;而对照组从第37周之后即显示机体活动功能持续降低。

在研探索.构画蓝图 SRP-9001是Sarepta公司与罗氏合作开发的DMD新药,旨在通过AAV将编码微型肌营养不良蛋白的基因传递到肌肉组织,使肌肉细胞表达对应的蛋白,从而减缓或阻止肌肉退化。SRP-9001已获得FDA快速通道认定,并获美国、欧盟、瑞士和日本的孤儿药认定。2022年11月,公司宣布FDA已受理该药物的生物制品许可申请(BLA),申请加速批准用于治疗非卧床的DMD患者,得益于丰富的临床前研究及多项临床试验的证据,SRP-9001显示稳定的安全性,并在治疗后的第1、2和4年评估中显示多个研究终点上的阳性。所有患者在第90天均证实了载体转导成功,NSAA评分显示功能改善、肌酸激酶(CK)水平降低。根据NSAA评分,患者的运动功能评分在随访2年后较基线增加7.0分。SRP-9001非常有希望成为针对致病基因根源、一次性治疗DMD的首个基因疗法。
PF-06939926是另一款在研的微型肌营养不良蛋白的基因疗法,由辉瑞公司研发,以重组腺相关病毒血清型9(rAAV9)衣壳为递送载体,具有靶向定位肌肉组织的潜力。早期对9名接受该药物治疗12个月的数据表明,微型肌营养不良蛋白数量显著增加,北极星动态评估(NSAA)得分显著提高,在糖皮质激素缓解支持的患者队列中,未出现严重不良事件。2021年1月,该药物的临床3期试验CIFFREO即已启动,预期在15个国家的55个临床试验中心入组99例稳定接受激素治疗的4-7岁门诊男性患者;直至同年12月Ib期临床试验的卧床队列发生了一例患者死亡,导致研究宣告暂停。由clinicaltrials.gov信息显示,2022年12月企业更新该药物的研究进展,Ib期临床试验未再招募患者,临床3期试验处于进行状态、招募中。
EDG-5506是罕见肌肉病治疗公司Edgewise公司在研的一种拟应用于治疗BMD、DMD和肢带型肌营养不良症(LGMD)的口服、小分子药物,被设计用来稳定快速收缩的骨骼肌纤维,通过抑制肌球蛋白来保护肌纤维免受损害。先前的临床前治疗已证明其疗效潜力,经长期治疗不但能延缓患者肌肉退化,还能改善心脏纤维化。2022年9月,Edgewise公布了EDG-5506 在BMD成人研究的4个月中期结果,肌酸激酶和快速骨骼肌肌钙蛋白I分别平均降低了29%和74%,北极星移动评价量表(NSAA)平均增加了1.17个点,这些数据突出了EDG-5506改变疾病进程的潜力。2022年10月,另有一项名为LYNX的安慰剂对照临床2期试验已经启动,用以进一步评估EDG-5506应用于DMD儿童的安全性、药代动力学及肌肉损伤相关生物标志物指标的变化;该试验预计纳入27例患者,接受12周的治疗,再进入长达9个月的开放标签研究阶段。
Vamorolone是一种是非泼尼松龙衍生物的类固醇药物,又称温和激素,由美国ReveraGen公司研发,瑞士的Santhera公司及中国的曙方医药均加入该药品上市推动与经营计划,2022年10月已完成向FDA滚动提交上市申请。其在保留激素(如泼尼松/泼尼松龙)抗炎效果的同时,减轻部分由传统激素带来的副作用,以此延长 DMD 患者的激素使用时间,进一步减缓病程。2022年10月,JAMA Neurology发表了该药与安慰剂、泼尼松龙对照的随机双盲临床试验结果,针对的是先前尚未接受过皮质类固醇治疗的DMD男性儿童患者,6mg剂量组在第6周即显示优于安慰剂组,直至12周达显著提升,并疗效维持至24周,非劣效于泼尼松龙组。而在治疗不良作用方面,如所预期,Vamorolone比泼尼松龙组在身高、骨骼不良影响上更小。



















