近半年来,宁宁的父母操碎了心。宁宁今年6岁了,6个月前她的理解记忆力突然减退,学习能力下降,原本掌握的生活技能也不会了,而且走路易跌倒。宁宁的父母十分焦急,带着孩子到当地医院就诊,颅脑MRI提示有肾上腺脑白质营养不良可能。为了救孩子,他们带着宁宁前往北京一家医院做了基因检测,确诊患上了罕见的遗传性疾病--X连锁肾上腺脑白质营养不良。
X连锁肾上腺脑白质营养不良到底是什么疾病?让我们走进这种罕见病。
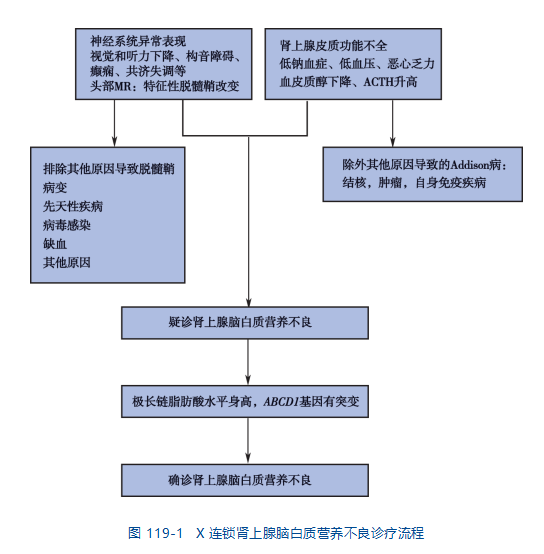
肾上腺脑白质营养不良(adrenoleukodystrophy,ALD)是X连锁隐性遗传病,是一种最常见的过氧化物酶体病,主要累及肾上腺和脑白质,半数以上的病人于儿童或青少年期起病,主要表现为进行性的精神运动障碍,视力及听力下降和(或)肾上腺皮质功能低下等。本病发病率约为0.5/10万~1/10万,95%是男性,5%为女性杂合子。无种族和地域特异性。
一般概述
X连锁肾上腺脑白质营养不良(ALD)是一种罕见的遗传性疾病,会影响神经系统的白质和肾上腺皮质。许多受影响的人在儿童期或成年期都经历过严重的神经系统问题,并伴有不同类型的残疾。一些受影响的个体还患有肾上腺功能不全,这意味着某些激素(如肾上腺素和皮质醇)的产生量减少,导致血压、心率、性发育和生殖异常。ALD 是一种 X 连锁隐性遗传病,由 ABCD1 基因的变异(突变)引起。因为它是一种 X 连锁疾病,男性比女性出现更严重的并发症,而一些女性则没有任何症状。ALD 可根据症状和发病年龄分为不同类型。
症状与体征
即使是同一家庭的成员,其体征和症状也可能有很大差异。有些人在婴儿期或儿童期有严重的并发症,而另一些人在成年后会出现症状。有些人直到成年才出现症状(无症状)。疾病的进展也可能有所不同。有不同形式的 ALD。典型 ALD 患者出现中枢神经系统损害和肾上腺皮质功能低下两类症状。神经系统症状和肾上腺皮质功能减退的症状可同时出现或相继出现。少数患者可单独出现。肾上腺皮质功能不全的表现轻重不一。根据发病年龄、受累部位、进展速度等临床表型的差异,ALD 分为 7 型,包括儿童脑型、青少年脑型、成人脑型、肾上腺脊髓神经病型(adrenomyeloneuropathy,AMN)、艾迪生型、无症状型和杂合子型。
1、儿童脑型 ALD
儿童脑型 最为常见,占所有 ALD 患者 35%。多在 4~8 岁发病,在3-10岁间出现神经系统症状。3岁前几乎不表现任何症状。
受影响的男性将正常发育,然后开始表现出先前获得的技能的丧失(退化)。在失去技能之前,受影响的男性可能会出现行为问题,包括注意力缺陷和多动障碍 (ADHD) 以及学习障碍。受影响的个体通常会出现认知缺陷,这意味着他们的心理过程可能受损并且难以获取信息和知识。这意味着受影响的儿童在学校的表现可能会下降。他们可能会在学校或不同时间“空出”,难以理解言语,难以阅读或理解书面文字,空间参考有困难,并表现出手写能力下降。
稍后,他们会出现其他症状,包括视力清晰度下降(视力下降)、听力丧失、步态困难,以及最终四肢无力和僵硬、抽搐或癫痫发作。最终,受影响的儿童失去了大部分神经功能,完全丧失了失明、耳聋和无法自主行动的能力。该疾病将进一步发展,导致植物人状态和死亡,通常在神经系统症状出现后的 2-3 年内。
2、艾迪生型
艾迪生型 占 ALD 患者的 10% ~14%,发病年龄在 2 岁至成年期。以原发性肾上腺皮质功能不全为主要表现,包括疲劳、意外体重减轻、恶心、呕吐、胃肠道问题、虚弱、早晨头痛、低血压(低血压)和低血糖水平(低血糖),许多受影响的男性可能会出现皮肤晒黑,包括未暴露在阳光下的区域(皮肤色素沉着)。而神经系统的异常多在中年后出现。
3、肾上腺髓质神经病 (AMN)型
AMN 型 约占 ALD 的 27%,20 岁后或中年起病,主要累及脊髓白质,周围神经病变较轻,无炎症性损伤。表现为下肢进行性痉挛性瘫痪、括约肌功能紊乱和性功能障碍。AMN 进展较慢。最初的症状通常是腿部进行性僵硬和无力(痉挛性截瘫)。受影响的男性可能会出现行走问题或以不寻常的方式行走(异常步态)。多发性神经病引起的麻木和疼痛也是常见症状。
由于括约肌功能障碍,受影响的男性还可能表现出勃起功能障碍和肠道和膀胱控制问题。括约肌是控制身体某些通道变窄或变宽的肌肉。尿道括约肌是两块肌肉,它们控制尿液从膀胱通过将尿液排出体外的细管(尿道)。尿道括约肌控制不佳会导致排尿功能障碍。此外,许多男性还会出现过早秃顶和头发稀疏的情况。
4、成人脑型 ALD占所有 ALD 病人的 2% ~4%,于 20 岁以后起病,颅内病变进展迅速, 无 AMN 表现。在所有受影响的男性中,至少有 20% 的人会出现认知能力下降,这与儿童脑型男孩所见的情况相似。这些患者会出现类似于儿童脑 ALD 的进行性神经系统症状,通常会导致严重的神经功能障碍,并最终导致植物人状态或死亡。
5、青少年脑型 占所有 ALD 病人的 4% ~7%,多于 10~20 岁起病,临床表现类似于儿童脑型,该型进展缓慢。
6、无症状型或Addison’s-only ALD,通过检查发现血极长链脂肪酸(VLCFA)升高或ABCD1 基因存在突变,但患者无相应的临床症状。
7. 杂合子型 ABCD1 基因突变的女性携带者,可能会出现轻度的 AMN 症状。随着年龄增加,临床表现更加明显。在 60 岁后,65%的杂合子患者会出现 AMN 临床表现,但症状轻微。很少出现脑部症状、周围神经病及肾上腺皮质功能减退。
大约 20% 的 40 岁以下女性 ALD 携带者会出现症状。到 60 岁时,这一比例达到 90% 左右。肾上腺功能不全和大脑受累在女性中很少见,但也可能发生。
另外,也有报道发现新的形式的,如脊髓小脑变异型,见:Neurology病例:X连锁肾上腺脑白质营养不良:脊髓小脑变异型
病因
该病由 X 染色体上(Xq28)ABCD1(adenosine triphosphate-binding cassette D1) 基因突变引起,导致过氧化物酶体功能缺失,长链脂肪酸不能被 b 氧化,大量极长链脂肪酸(very long chain fatty,VLCFA) 沉积于大脑白质和肾上腺,导致神经系统功能异常及肾上腺皮质功能减退。95%的患者为男性,而女性多为杂合子,属于疾病基因突变的携带者。
在肾上腺皮质中,极长链脂肪酸的异常积累与产生激素的细胞死亡有关,而确切的机制尚不清楚。肾上腺皮质的损伤也可能是由于免疫系统对脂肪积累的异常反应造成的。
在一些被称为杂合子的女性中,她们遗传了 ALD 疾病基因的一个拷贝,X 染色体上的疾病特征可能并不总是被另一个 X 染色体上的正常基因所掩盖。因此,这些女性可能会表现出与 ALD 相关的症状。
鉴别诊断
以下疾病的症状可能与 ALD 的症状相似。比较可能有助于鉴别诊断。具体的鉴别诊断取决于 ALD 的具体形式。
儿童脑 ALD 需要与其他形式的脑白质营养不良症区分开来,例如 Krabbe 病或芳基硫酸酯酶 A 缺乏症、莱姆病、多发性硬化症、注意力缺陷多动障碍和各种脑肿瘤。肾上腺脊髓神经病需要与多发性硬化、遗传性痉挛性截瘫、肌萎缩侧索硬化和脊髓肿瘤相鉴别。
诊断
ALD 的诊断基于特征性症状的识别、详细的患者和家族史、全面的临床评估和各种专业测试。
一些婴儿可以通过新生儿筛查得到诊断。新生儿筛查是一种特殊类型的筛查测试,新生儿接受检查他们是否患有某些疾病。这种筛查主要是通过检查干血斑来完成的。因为新生儿筛查是一项筛查测试,阳性结果并不意味着婴儿肯定患有疾病。通常,必须进行重复测试以确认诊断。如果新生儿筛查呈阳性,则可能需要进行基因检测以确定导致 ALD 的特定基因变化(突变)。
2016 年,ALD 被添加到美国推荐的统一新生儿筛查小组 (RUSP) 中。但是,每个州都确定该州内的新生儿筛查计划中包括哪些特定疾病。截至 2019 年 10 月,只有十几个州通过新生儿筛查检测 ALD,尽管更多州计划将这种疾病添加到检测项目中。
临床测试和检查
最初的诊断测试通常是血液测试,以测量血浆中超长链脂肪酸的水平。如果这些水平特别高,或者血液中这些脂肪分子的比例不正常,医生将下令进行基因检测以确认诊断。一些携带 ALD 的女性血液中的长链脂肪酸含量正常。怀疑患有这种疾病的女性可能需要进行基因检测以明确排除诊断。
分子遗传学检测可以确诊。分子遗传学检测可以检测已知会导致 ALD 的 ABCD1 基因突变,但只能在专业实验室作为诊断服务使用。
医生还将通过一种称为 ACTH 刺激测试的测试来测试肾上腺的功能。ACTH 代表促肾上腺皮质激素,由垂体产生。ACTH浓度的增加导致肾上腺激素的产生增加。具体来说,血浆中的皮质醇应该会升高。
可能会推荐一种称为磁共振成像 (MRI) 的专门成像技术来评估 ALD 如何影响大脑。MRI 使用磁场和无线电波来生成特定器官和身体组织(包括大脑)的横截面图像。这使医生可以查看大脑是否已受损,包括大脑白质中髓鞘的丢失。
治疗
治疗可能需要专家团队的协调努力。儿科医生、普通内科医生、专门诊断和治疗大脑和神经系统疾病的医生(小儿神经科医生)、成人神经科医生、专门诊断和治疗泌尿系统疾病的医生(泌尿科医生)、专门诊断和治疗的医生内分泌系统疾病(内分泌学家)、精神科医生、物理治疗师和其他医疗保健专业人员可能需要系统和全面地计划治疗。
建议对受影响的个人及其家人进行遗传咨询。对整个家庭的社会心理支持也很重要。
应密切监测没有症状(无症状)的男孩是否有脑部疾病迹象。这个群体往往是通过新生儿筛查发现的男孩,或者是因为以前受影响的家庭成员而被早期诊断出来的。造血干细胞移植等治疗应仅适用于尚未出现神经系统症状的 MRI 异常变化的男孩。
对于肾上腺功能不全的人,皮质类固醇替代疗法是必不可少的。脊髓疾病、泌尿系统并发症、多发性神经病的管理往往遵循常规或标准指南。
在对婴儿或儿童进行初步诊断后,可能会进行神经发育评估,受影响的男孩需要在常规基础上进行重复的 MRI 研究以进行疾病监测。尽早识别脑部 MRI 变化很重要,因为在神经系统症状之前有早期 MRI 变化的个体在接受治疗时具有最佳结果。应向所有儿童和成人提供定期重新评估和调整服务。可能需要额外的医疗、社会和/或职业服务,包括专业学习计划。
异基因造血干细胞移植 (HSCT) 用于治疗某些个体,特别是有中枢神经系统受累证据且处于疾病早期且没有神经系统症状的年轻男孩或青少年,是目前的护理标准,并且只有现有的有效治疗以阻止儿童期神经系统症状的进展。
造血干细胞是在骨髓中发现的特殊细胞,可以生长或成熟为不同类型的细胞。在同种异体干细胞移植中,受影响的个体接受来自健康人(称为供体)的造血干细胞。在过去的二十年中进行的一系列研究表明,HSCT 可以阻止 ALD 中神经系统疾病的进展,尽管它不能改善肾上腺功能不全。HSCT 是一项具有重大风险的主要医疗程序。由于异基因 HSCT 仅在疾病的早期阶段有效,因此将新生儿筛查扩大到所有州以及所有被诊断患有 ALD 的男孩都接受重复的脑部 MRI 研究并定期与神经科医生进行随访是相关的。
研究中的治疗方法
HSCT 也正在被研究作为一种潜在的治疗方法,用于治疗因 ALD 导致严重残疾的受影响成年人。仅对少数受影响的个体进行了研究。需要更多的研究来确定 HSCT 对患有 ALD 的成年人的长期安全性。
基因疗法也正在被研究作为另一种治疗受影响个体的方法。在基因治疗中,患者体内存在的缺陷基因被正常基因取代,以产生活性酶并防止相关疾病的发展和进展。鉴于能够在所有疾病部位产生工作蛋白的正常基因的永久转移,这种治疗形式理论上最有可能导致“治愈”。正在进行初步研究,以确定基因治疗对某些 ALD 患者的长期安全性和有效性。Lenti-D慢病毒载体基因治疗可有效缓解早期X-连锁肾上腺脑白质营养不良患者的病情,是异基因造血干细胞移植之外的一个治疗选择。
见:NEJM:早期X-连锁肾上腺脑白质营养不良的基因治疗
Lorenzo油(Lorenzo’s oil)是 ALD 的实验性治疗方法。研究表明,在一些儿童中,洛伦佐油降低了尚未出现任何症状(无症状)的儿童出现神经系统症状的风险。然而,它对已经表现出这种疾病的神经系统症状的儿童无效。一般来说,像洛伦佐油这样的饮食调整或限制脂肪食物的摄入量并未显示出临床有效性,仍处于研究阶段。


















